gff/gtf格式
1)gff3及gtf2简介
一个物种的基因组测序完成后,需要对这些数据进行解读,首先要先找到这些序列中转录起始位点、基因、外显子、内含子等组成元件在染色体中的位置信息(即注释)后才能再进行深入的分析。gff/gtf是贮存这些注释信息的两种文件格式。
GFF(general feature format):这种格式主要是用来注释基因组。 现大部分利用的是第三版,即gff3。
GTF(gene transfer format):主要是用来对基因进行注释。当前所广泛使用的gtf格式为第二版,即gtf2 。
1.1)GFF3
GFF3允许使用#作为注释符号 ,除去注释外,主体部分共有9列。 GFF3中每一列的含义:seqid source type start end score strand strand attributes
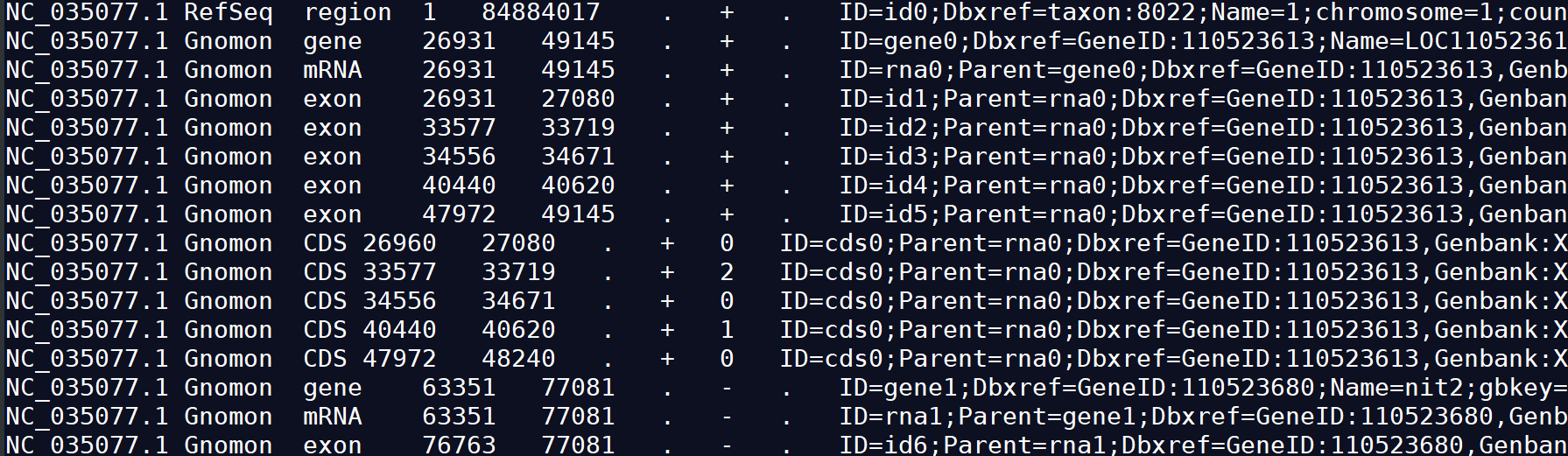
1) seqid :序列的id。(The name of the sequence where the feature is located.)
2)source:注释的来源,一般指明产生此gff3文件的软件或方法(e.g. Augustus or RepeatMasker)。如果未知,则用点(.)代替。
3)type: 类型,此处不受约束,但为下游分析方便,建议使用gene,repeat_region,exon,CDS,或SO对应编号等。
4)start:起始位置,从1开始计数(区别于bed文件从0开始计数)。
5)end:终止位置。
6)score:得分,注释信息可能性说明,可以是序列相似性比对时的E-values值或者基因预测是的P-values值。”.”表示为空。(indicates the confidence of the source on the annotated feature)
7)strand:“+”表示正链,“-”表示负链,“.”表示不需要指定正负链,“?” 表示未知.
8)phase :步进。仅对编码蛋白质的CDS有效,本列指定下一个密码子开始的位置。可以是0、1或2,表示到达下一个密码子需要跳过碱基个数。
9)attributes:属性。一个包含众多属性的列表,格式为“标签=值”(tag=value),不同属性之间以分号相隔。
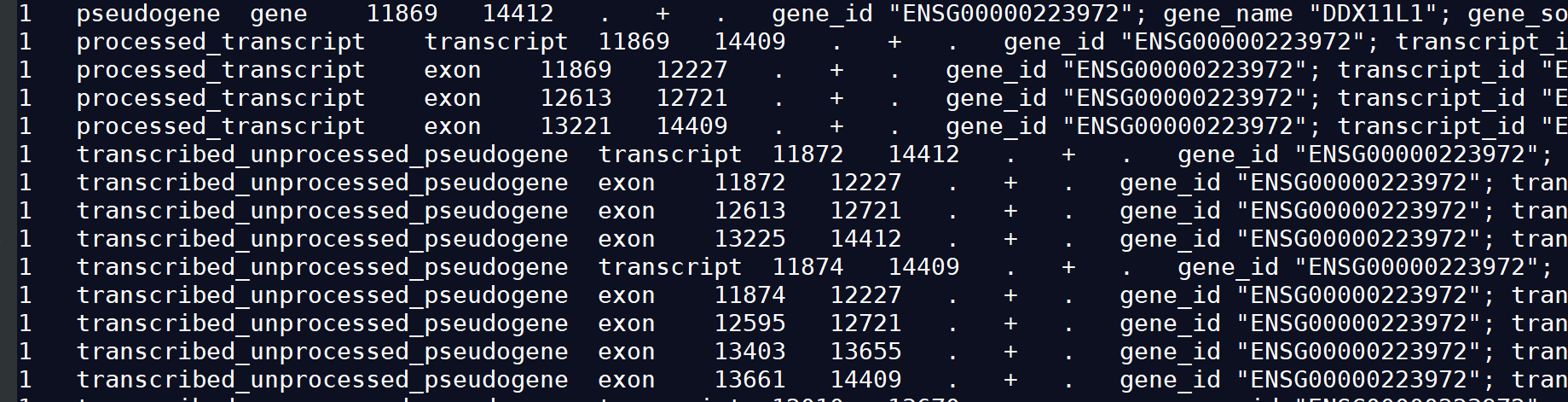
1.2)GTF2
gtf文件也是由9列组成,其中每一列含义:seqname source feature start end score strand frame attributes
1) seqname: 序列的名字。通常格式染色体ID或是contig ID。
2) source:注释的来源。通常是预测软件名或是公共数据库。
3) start:起始位置,从1开始计数。
4) end:终止位置。
5) feature :基因结构.根据所使用软件不同,feature types必须注明。CDS,start_codon,stop_codon是一定要含有的类型。
6) score :这一列的值表示对该类型存在性和其坐标的可信度,不是必须的,可以用点“.”代替。
7) strand:链的正向与负向,分别用加号+和减号-表示。
8) frame:密码子偏移,可以是0、1或2。
9) attributes:必须要有以下两个值:
gene_id value: 表示转录本在基因组上的基因座的唯一的ID。gene_id与value值用空格分开,如果值为空,则表示没有对应的基因。
transcript_id value: 预测的转录本的唯一ID。transcript_id与value值用空格分开,空表示没有转录本。
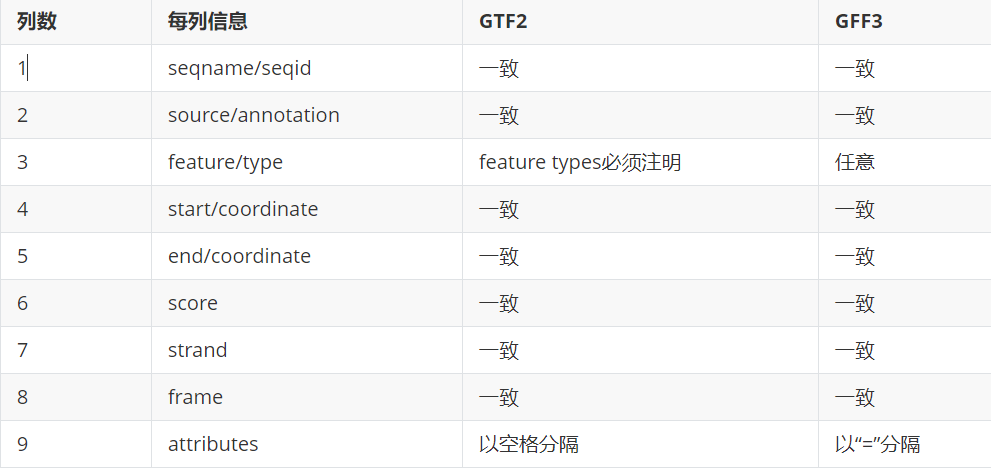
2)GFF3和GTF2之间的异同及相互转换。
---------------------------------------------------
GFF3和GTF2之间的转换可以用Cufflinks里面的工具"gffread":
gffread my.gff3 -T -o my.gtf #gff2gtf
gffread merged.gtf -o- > merged.gff3 #gtf2gff
3) 习题
---------------------------------------
3.1)gff3格式的功能是是什么?目前有几版?
3.2)gff3共有多少列?每一列的含义是什么?
3.3)gff3中的第8列代表的0,1,2分别代表什么含义?
3.4)gff3第9列不同属性之间是用什么符号分割的?
3.5)gtf2和gff3格式上有何异同?
3.6)gtf2和gff3在功能上有什么差异?
3.7)gtf2第9列中不同属性用什么符号分割?
3.8)如何将gtf和gff之间相互转换?
3.9)统计test.gff文件中组装出来的染色体条数
3.10)统计test.gff文件中lnc_RNA个数
3.11)统计基因组文件test.gff中有多少个基因
3.12)求最长基因的长度
3.13)查找一个基因下有3个转录本的基因个数
3.14)求相位为2的cds个数
3.15)找出基因含有最多的外显子的个数
3.16) 将test.gff转化为test.gtf
3.17)统计test.gtf中transcript的个数
3.18)根据test.gtf统计位于正链上的exon的个数
3.19)将test.gtf中所有的gene ID都统计出来
3.20) 找出test.gtf中位于正链上的最长的基因
4) 参考资源
---------------------------------------
https://en.wikipedia.org/wiki/General_feature_format
http://boyun.sh.cn/bio/?p=1602
gff/gtf格式的更多相关文章
- 探索gff/gtf格式
参考: GFF格式说明 Generic Feature Format Version 3 (GFF3) 先下载一个 gtf 文件浏览一下 1 havana gene 11869 14409 . + . ...
- (转) gffcompare和gffread | gtf | gff3 格式文件的分析 | gtf处理 | gtfparse
工具推荐:https://github.com/openvax/gtfparse 真不敢相信,Linux自带的命令会这么强大,从gtf中提取出需要的transcript,看起来复杂,其实一个grep就 ...
- GTF/GFF文件的差异及其相互转换
我们在做生物分析的时候,经常会碰到GFF格式的文件以及GTF格式的注释文件.他们有着相似的名字,甚至连内容都极为相似~那么,他们究竟差在哪里呢? GFF全称为general feature forma ...
- 关于基因组注释文件GTF的解释
GTF文件的全称是gene transfer format,主要是对染色体上的基因进行标注.怎么理解呢,其实所谓的基因名,基因座等,都只是后来人们给一段DNA序列起的名字而已,还原到细胞中就是细胞核里 ...
- 如何使用SnpEff 对SNP结果进行分析
SnpEff is a variant annotation and effect prediction tool. It annotates and predicts the effects of ...
- 【转录组入门】6:reads计数
作业要求: 实现这个功能的软件也很多,还是烦请大家先自己搜索几个教程,入门请统一用htseq-count,对每个样本都会输出一个表达量文件. 需要用脚本合并所有的样本为表达矩阵.参考:生信编程直播第四 ...
- tophat的用法
概述:tophat是以bowtie2为核心的一款比对软件. tophat工作分两步: 1.将reads用bowtie比对到参考基因组上. 2.将unmapped-reads打断成更小的fragment ...
- Augustus指南(Trainning部分)
Augustus指南 官方 Tutorial Index Augustus是一个真核生物基因预测软件,目前有网页服务端和本地版,它基于Hidden-Markov Model(隐马尔科夫链模型HMM)( ...
- bedtools 每天都会用到的工具
详细的使用说明:http://bedtools.readthedocs.org/en/latest/ Collectively, the bedtools utilities are a swiss- ...
随机推荐
- BASIC-6_蓝桥杯_杨辉三角形
示例代码: #include <stdio.h>#include <stdlib.h> int main(void){ int n = 0 ; int i = 0 , j = ...
- maven学习(6)-Maven依赖范围
一.maven依赖范围: classpath 分为三种:编译classpath , 测试classpath , 运行classpath Scope 选项如下: Compile:编译依赖范围.默认就是c ...
- 基于jQuery.i18n.properties实现前端网站语言多版本
我是参考播客做了个demo:http://blog.csdn.net/aixiaoyang168/article/details/49336709 jQuery.i18n.properties采用.p ...
- [UE4]roll pitch yaw
UE4中的定义: 一.Roll,绕着X轴旋转的角度 二.Pitch,绕着Y轴旋转的角度 三.Yaw,绕着Z轴旋转的角度 Rotator 一.(Roll,Pitch,Yaw) 二.Rotator(0,0 ...
- Java 泛型小结
1.什么是泛型? 泛型(Generics )是把类型参数化,运用于类.接口.方法中,可以通过执行泛型类型调用 分配一个类型,将用分配的具体类型替换泛型类型.然后,所分配的类型将用于限制容器内使用的值, ...
- php while循环控制实例讲解
while循环是PHP中最简单的循环,其基本格式为: while (expr){ statement } 或者 while (expr): statement endwhile; 该语法表示,只要ex ...
- ACM MM | 中山大学等提出HSE:基于层次语义嵌入模型的精细化物体分类
细粒度识别一般需要模型识别非常精细的子类别,它基本上就是同时使用图像全局信息和局部信息的分类任务.在本论文中,研究者们提出了一种新型层次语义框架,其自顶向下地由全局图像关注局部特征或更具判别性的区域. ...
- 如何对hashmap按value值排序
http://bbs.csdn.net/topics/90321713 这个帖子中没有我想要的答案,treemap是根据key排序的,想以value排序,那么可以key,value互换一下,不过这样的 ...
- Fork-Join 原理深入分析(二)
本文是将 Fork-Join 复杂且较为庞大的框架分成5个小点来分析 Fork-Join 框架的实现原理,一个个点地理解透 Fork-Join 的核心原理. 1. Frok-Join 框架的核心类 ...
- Java集合入门
内容: 1.认识集合 2.Iterator迭代器 1.认识集合 (1)什么是集合 前面的学习,我们知道数据多了,使用数组存放.而且数组中存放的都是基本类型的数据,并且数组是定长的. 当在程序中创建的对 ...