SAGE|DNA微阵列|RNA-seq|lncRNA|scripture|tophat|cufflinks|NONCODE|MA|LOWESS|qualitile归一化|permutation test|SAM|FDR|The Bonferroni|Tukey's|BH|FWER|Holm's step-down|q-value|
生物信息学-基因表达分析
为了丰富中心法则,研究人员使用不断更新的技术研究lncRNA的方方面面,其中技术主要是生物学上的微阵列芯片技术和表达数据分析方法,方方面面是指lncRNA的位置特征。
Background:根据中心法则,发现DNA与RNA与protein之间的关系,此时认为找到的RNA全部用于编码protein,但是实验结果中:非编码RNA含量高,而coding区只占很少的一部分。研究非编码RNA,发现noncoding与protein expression有关,所以总思路变成了研究noncoding区从而丰富中心法则,而研究noncoding区的前提是转录组分析。
转录组研究
1.依据实际研究的问题,可以有不同的思路
时间(生长发育不同阶段)特异性
空间(身体不同区域)特异性
Normal 与否
- 随着测序技术的发展,出现了以下RNA的测序手段
SAGE:将RNA反转录得到的cDNA打碎,利用sanger测序法得到最后的序列
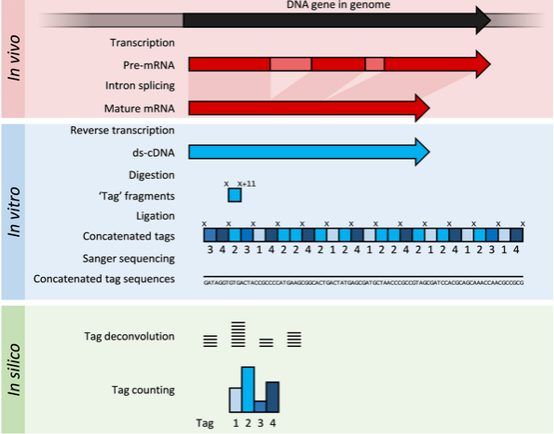
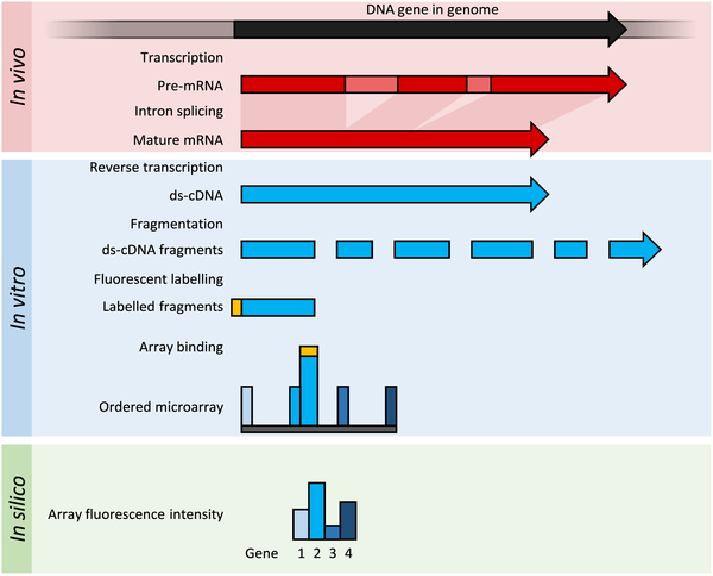
DNA微阵列: 将RNA打碎,用基于reference的探针(特指基因芯片)测得序列,此方法灵敏性高
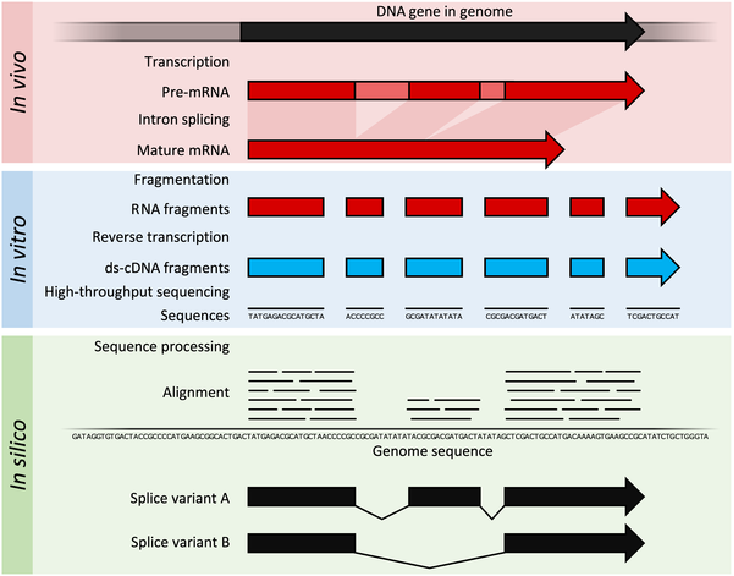
RNA-seq:将RNA打碎,再将片段RNA反转录cDNA,利用二代测序(short reads)assembly而成。
Long noncoding RNA
定义:
1.因为200bp,所以long;
2.因为No protein produce但同时有与coding gene具有相似结构(都有intro和exon),所以lncRNA长期被认为是假基因,直到发现其和转录调控有关系,才开始被重视。
起源:
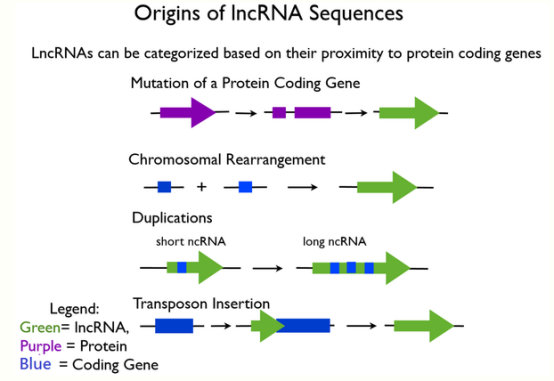
- coding gene mutation变成noncoding gene,所以无法coding protein
- Chromosome重组
- sncRNA加倍成为lncRNA
- sncRNA插入coding gene使之丧失功能但长度变长,成为lncRNA
分类:基于与protein的位置

lncRNA发现历史:
在大规模测序出现之前,只能利用sanger发现单个lncRNA。在完成human genome 之后,基于该数据库,得到的芯片,大大加速了lncRNA的发现。随着二代测序的大规模使用,大量发现结构,因此诞生了基于不同算法的assembly&annotation软件(eg,scripture、tophat、cufflinks)。所以,如今的思路是利用不同工具处理相同数据,将得到的不同结果集成为更准确的genome,从而得到比较可靠的reference。(review:Rinn and Chang,2012)但是2015用更多的原始数据用同样方法重做,发现其中有79%之前没有的,这是因为原始数据采集更在时间和空间上更为细致了。如今,得到了NONCODE (数量最多dataset)、MITranscript (最新)等各有侧重的数据库。
Assembly比对方式:
1.交叉--保留
2,存在不交叉的,利用其它参数信息(eg,位置信息)筛选
Gene expression 分析:
原则:重复&随机&间隔
Replication:biology(eg,同一个gene测100次得到100个sequence信息) &technical(对于100个sequence信息,可以随机抽取其中的任意N个,这种抽取做M次),这都是为了数据能更好的反应客观事实。
Randomization:无论是抽选或是物理设备参数设定,都要保证随机性。
Blocking:生物学实验的连贯性(物理条件一致,eg,一天一个完整实验,而不是一天所有完整实验中的一个步骤)
以微阵列方法为例的流程:
在仪器上得到荧光信号,将这些应该信号按照光的亮度赋值,由图转变为表,该表就是表达谱。根据荧光信号的特点,比如中间较强周围较弱就比较好,进行质量控制,也就是筛选质量较好的sample。此时,所有gene的表达量都在一个表格里面,其中使用三张芯片就被叫做生物学重复,以此创造:
|
芯片1 |
芯片2 |
芯片3 |
|
|
Gene1 |
2 |
4 |
4 |
|
Gene2 |
5 |
4 |
14 |
|
Gene3 |
4 |
6 |
8 |
可知芯片1比芯片3的光照程度整体普遍弱,这可能是由于物理因素造成的。为了进行比较,要将其数值进行归一化(normalization),依据比价对象的不同,可将方法分为两种:
- Intra:eg,芯片1内部比较不同gene表达量(技术是双通道的MA,归一化方法是LOWESS )
- Inter:eg,gene1在不同芯片上的表达量,(技术是单通道;思路可以是中位数归一化或qualitile归一化图
其中,qualitile归一化 的过程是:
|
芯片1 |
芯片2 |
芯片3 |
|
|
Gene1 |
2 |
4 |
4 |
|
Gene2 |
5 |
4 |
14 |
|
Gene3 |
4 |
6 |
8 |
不看属于哪个gene,在芯片内部进行排序:
|
芯片1 |
芯片2 |
芯片3 |
|
|
Mean=10/3 |
2(Gene1) |
4(Gene1) |
4(Gene1) |
|
Mean=16/3 |
4(Gene3) |
4(Gene2) |
8(Gene3) |
|
Mean=25/3 |
5(Gene2) |
6(Gene3) |
14(Gene2) |
求均值并写回去,这样做是为了避免物理错误,物理错误会导致整张芯片的效果都不好。不用考虑排序会因为表达量一致而造成误差,因为实际上不存在表达量完全一致的情况:
|
芯片1 |
芯片2 |
芯片3 |
|
|
Gene1 |
10/3 |
10/3 |
10/3 |
|
Gene2 |
25/3 |
16/3 |
25/3 |
|
Gene3 |
16/3 |
25/3 |
16/3 |
归一化之后,利用假设检验证明实际问题。
当数据服从某种分布(既有参数)时,可就某些参数(eg,mean)进行假设检验
当数据未知分布(即无参数),则采用permutation test:
使用的理论是t检验中的SAM,因为t-test需要有参数,但是现在数据未知参数,所以加入置换测试的方法
SAM"Statistical Analysis of Microarrays" specifically designed for microarray data analysis. It relies on the non-parametric permutation test. SAM is a variant of the t-test. Shown below is an excerpt of Tushner's paper describing the SAM algorithm:

Eg,分为测试组(treatment)和对照组(control):下图是它们的数据分布D
|
Key |
T1 |
T2 |
T3 |
C1 |
C2 |
C3 |
|
Gene1 |
1 |
2 |
3 |
4 |
5 |
6 |
将其随机打乱成1000个类似于下方表格的sample:
比如其中的2个是:
|
Key |
T1 |
T2 |
T3 |
C1 |
C2 |
C3 |
Mean of T |
Mean of C |
STDEV of all |
|
Gene1 |
6 |
1 |
2 |
3 |
4 |
5 |
3 |
4 |
1.870828693 |
|
Key |
T1 |
T2 |
T3 |
C1 |
C2 |
C3 |
Mean of T |
Mean of C |
STDEV of all |
|
Gene1 |
5 |
1 |
2 |
3 |
4 |
6 |
2.666666667 |
4.333333333 |
1.870828693 |
其中,S0是给定值,d可以认为是改良版的t值,如此得到1000个t值,这1000个t值可构成正态分布统计图表,最后根据总体分布,找到数据分布D的p值,而后根据该p值判断假设检验的结果。
由此,可以得到所有基因对应的p值,但是因为p值的错误率很高,所以需要采用多重假设检验对p值做检查。
可采用FDR
首先对于每个基因来说,都有p值,p值的含义是false positive rate(FPR,假正率):q值是false discovery rate (FDR):
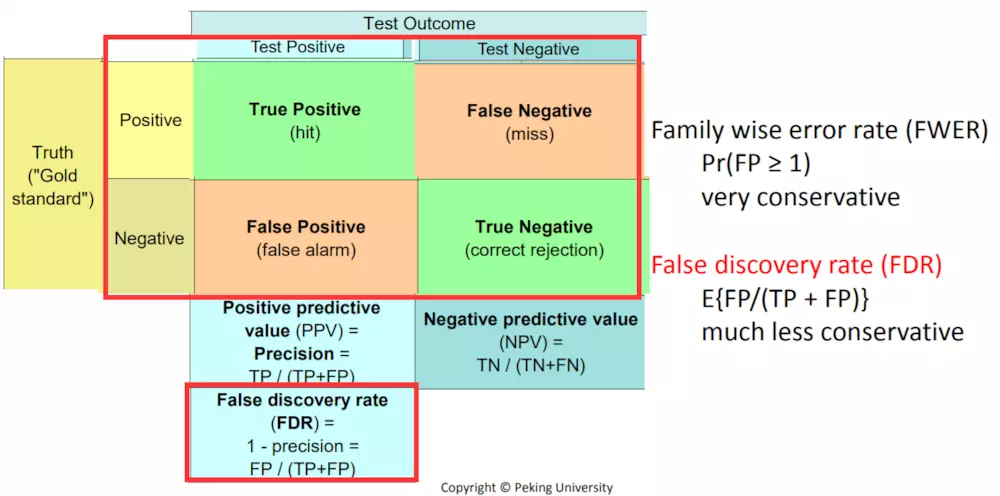
因为未知真实情况,但是从上图可知真实情况和估算情况之间的关系,所以可以使用别的方法计算FWER和FDR。
Control of FWER:
The Bonferroni procedure
Tukey's procedure
Holm's step-down procedure
Control of FDR
Benjamini–Hochberg procedure
Benjamini–Hochberg–Yekutieli procedure
例如,BH:
,
,所以q-value的期望即是FDR,所以,求出每个gene的q-value即可
原先每个gene都有自己的p值:
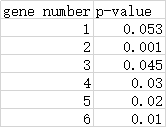
可靠gene的p值满足: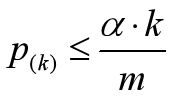
可靠gene的q值满足:
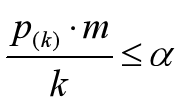
其中,k是gene number号,a是自己设定的0.05,m是所有gene个数,在这里是6;
把gene按照p值排序并据公式处理,得到:

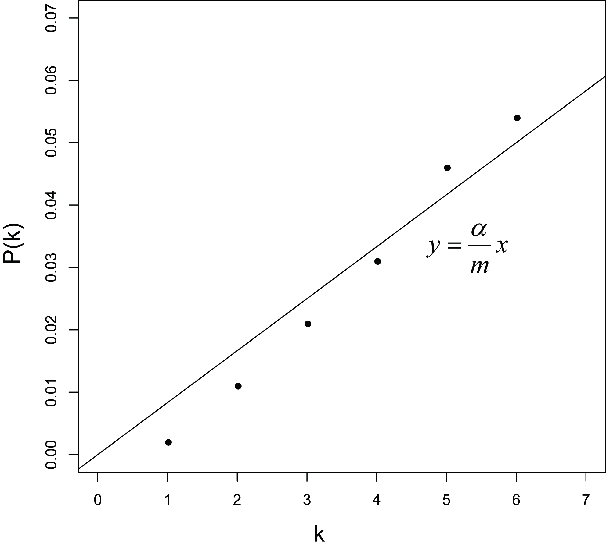
若写为函数表达则可得到上图,有四个gene的p值是可靠的。
SAGE|DNA微阵列|RNA-seq|lncRNA|scripture|tophat|cufflinks|NONCODE|MA|LOWESS|qualitile归一化|permutation test|SAM|FDR|The Bonferroni|Tukey's|BH|FWER|Holm's step-down|q-value|的更多相关文章
- 2. Transcribing DNA into RNA
Problem An RNA string is a string formed from the alphabet containing 'A', 'C', 'G', and 'U'. Given ...
- 长链非编码RNA(lncRNA)
长链非编码RNA(lncRNA) 转自:http://blog.sina.com.cn/s/blog_909da11301010bkz.html 长链非编码RNA(lncRNA)是一类转录本长 ...
- RNA seq 两种计算基因表达量方法
两种RNA seq的基因表达量计算方法: 1. RPKM:http://www.plob.org/2011/10/24/294.html 2. RSEM:这个是TCGAdata中使用的.RSEM据说比 ...
- RNA -seq
RNA -seq RNA-seq目的.用处::可以帮助我们了解,各种比较条件下,所有基因的表达情况的差异. 比如:正常组织和肿瘤组织的之间的差异:检测药物治疗前后,基因表达的差异:检测发育过程中,不同 ...
- 链终止法|边合成边测序|Bowtie|TopHat|Cufflinks|RPKM|FASTX-Toolkit|fastaQC|基因芯片|桥式扩增|
生物信息学 Sanger采用链终止法进行测序 带有荧光基团的ddXTP+其他四种普通的脱氧核苷酸放入同一个培养皿中,例如带有荧光基团的ddATP+普通的脱氧核苷酸A.T.C.G放入同一个培养皿,以此类 ...
- 使用Tophat+cufflinks分析差异表达
使用Tophat+cufflinks分析差异表达 2017-06-15 19:09:43 522 0 0 使用TopHat+Cufflinks的流程图 序列的比对是RNA分析 ...
- 02 Transcribing DNA into RNA
Problem An RNA string is a string formed from the alphabet containing 'A', 'C', 'G', and 'U'. Given ...
- tophat cufflinks cuffcompare cuffmerge 的使用
Cole Trapnell said: there are three strategies: 1) merge bams and assemble in a single run of Cuffli ...
- lncRNA研究
------------------------------- Long noncoding RNAs are rarely translated in two human cell lines. ( ...
随机推荐
- while read line do done < file
zzx@zzx120:~/test1$ cat file.txt 1122zzx@zzx120:~/test1$ cat ./read.sh #!/bin/bashwhile read line ...
- 安装使用离线版本的维基百科(Wikipedia)
1 相关背景 平常大家在上网查询一些基本概念的时候常常会参考维基百科上面的资料,但是由于方校长研制的GFW(长城防火墙系统)强大的屏蔽功能,好多链接打开以后,不出意外会出现著名的“404NOT FOU ...
- 吴裕雄--天生自然 JAVA开发学习:条件语句
public class Test { public static void main(String args[]){ int x = 10; if( x < 20 ){ System.out. ...
- [原]排错实战——通过对比分析sysinternals事件修复程序功能异常
原调试debug排错troubleshootprocess monitorsysinternals 缘起 最近,我们程序的某个功能在一台机器上不正常,但是在另外一台机器上却是正常的.代码是同一份,vs ...
- 谷歌发布自家物理密匙TitanKey,能让人们远离互联网“黑洞”吗?
随着隐私泄露事件的飞速增多,人们对如何保护自身隐私安全也越来越重视.而基于这种诉求,众多企业也在持续发力安全层面.毕竟,在目前危机四伏的互联网大环境下,真正能保护人们隐私安全的服务.设备等都是&quo ...
- Linux-exec族函数
1.为什么需要exec族函数 (1).fork子进程是为了执行新程序(fork创建子进程后,子进程和父进程同时被OS调度执行,因此子程序可以单独的执行一个程序,这样程序宏观上将会和父进程程序同时进行) ...
- C语言 指针在函数传参中的使用
int add(int a, int b) //函数传参的时候使用了int整型数据,本身是数值类型.实际调用该函数时,实参将自己拷贝一份,并将拷贝传递给形参进行运算.实参自己实际是不参与运算的.所 ...
- Python这五个坑,80%你不知道(对的,五个你知道1个就达到一般水平了)
1 含单个元素的元组 Python中有些函数的参数类型为元组,其内有1个元素,这样创建是错误的: c = (5) # NO! 它实际创建一个整型元素5,必须要在元素后加一个逗号: ! c = (5,) ...
- HTTP编码
HTTP编码 不仅仅URL需要编码,HTTP header也需要编码,HTTP body 无特殊要求 一般采用百分号编码:比如一个字节的ascii码值是 0x89 那使用百分号编码之后 输出是 %89 ...
- Opencv笔记(五)——把鼠标当画笔
学习目标: 学习使用 OpenCV 处理鼠标事件 学会使用函数cv2.setMouseCallback() 简单演示: 首先我们来创建一个鼠标事件回调函数,但鼠标事件发生是他就会被执 ...