【转录组入门】3:了解fastq测序数据
操作:需要用安装好的sratoolkit把sra文件转换为fastq格式的测序文件,并且用fastqc软件测试测序文件的质量
作业:理解测序reads,GC含量,质量值,接头,index,fastqc的全部报告,搜索中文教程
具体步骤
【1】SRA文件转换成fastq文件
-----单个文件转换
fastq-dump --gzip --split- -O outputdir -A file1.sra
-----多个文件批量转换
# 、编写一个脚本 sra_to_fq.sh
for I in `seq `
do
fastq-dump --gzip –split- -O ./fastq/ -A SRR35899${I}.sra
done # --split-:如果是双端测序数据,则输出两个文件,如果不是则只输出一个文件
# --gzip:输出格式为gzip的压缩文件(fastqc软件可以直接识别gzip压缩的文件)
# -A:accession序列号,输入的文件
# -O:outdir输出文件夹,指定输出路径 # 、运行脚本
bash sra_to_fq.sh
【2】QC(测序质量分析):多个文件批量进行
$ fastqc -q -t -o ./fastqc_result/ *.fastq.gz &
# -t :调用8个核心
# -q :安静运行,在运行过程中不会生成报告,只会在结束时将报告生成一个文件
# -o ../FastQC_result.raw/ :文件输出位置,输出到当前文件夹下的FastQC_result 子目录中
# *. fq.gz:,输入文件:当前目录下所有名字中有“ .fq.gz ”的文件
【3】查看QC结果
1、单个查看:鼠标双击打开html文件查看
2、批量查看:使用 moltiqc软件: moltiqc *fastqc.zip
Fastqc结果报告关注重点:
1).basic statistics
2).per base sequence quality
3).per base sequcence content
4).adaptor content
5).sequence duplication levels
主要的几个指标是GC含量,Q20和Q30的比例以及是否存在接头(adaptor)、index以及其他物种序列的污染等。
质控软件:
测序数据去掉接头:cutadapt
删掉测序质量差的reads:fastx_trimmer
理论知识
高通量测序之所以能够能够达到如此高的通量的原因就是他把原来几十M,几百M,甚至几个G的基因组通过物理或化学的方式打算成几百bp的短序列,然后同时测序。

在测序过程中,机器会对每次读取的结果赋予一个值,用于表明它有多大把握结果是对的。从理论上都是前面质量好,后面质量差。并且在某些GC比例高的区域,测序质量会大幅度降低。
因此,我们在正式的数据分析之前需要对分析结果进行质控
fastq格式
Fastq格式是一种基于文本的存储生物序列和对应碱基(或氨基酸)质量的文件格式。最初由桑格研究所(Wellcome Trust Sanger Institute)开发出来,现已成为存储高通量测序数据的事实标准。
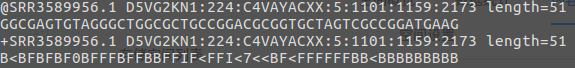
每条read由4行字符构成:
第一行:必须以@开头,后面跟着序列的唯一ID以及相关说明内容。
第二行:核酸序列,是有ATCGN字符组成。
第三行:“ ”开头,内容和第一行@后面的一样。
第四行:每个测序碱基质量,是用ASCII码来表示的,与第二行的字符数一致。
碱基质量得分与错误率的换算关系: Q = -10log10p(p表示测序的错误率,Q表示碱基质量分数)
ASCII值与碱基质量得分之间的关系: Phred64 Q=ASCII转换后的数值-64 Phred33 Q=ASCII转换后的数值-33
第三行质量序列格式
目前illumina使用的碱基质量格式为phred+33, 和Sanger的质量基本一致。
| Name | ASCII character range | Offset | Quality score type | Quality score range |
| Sanger, Illumina (versions 1.8 onward) | 33–126 | 33 | PHRED | 0–93 |
| Solexa, early Illumina (before 1.3) | 59–126 | 64 | Solexa | 5–62 |
| Illumina (versions 1.3–1.7) | 64–126 | 64 | PHRED | 0–62 |
质控结果解读
总览
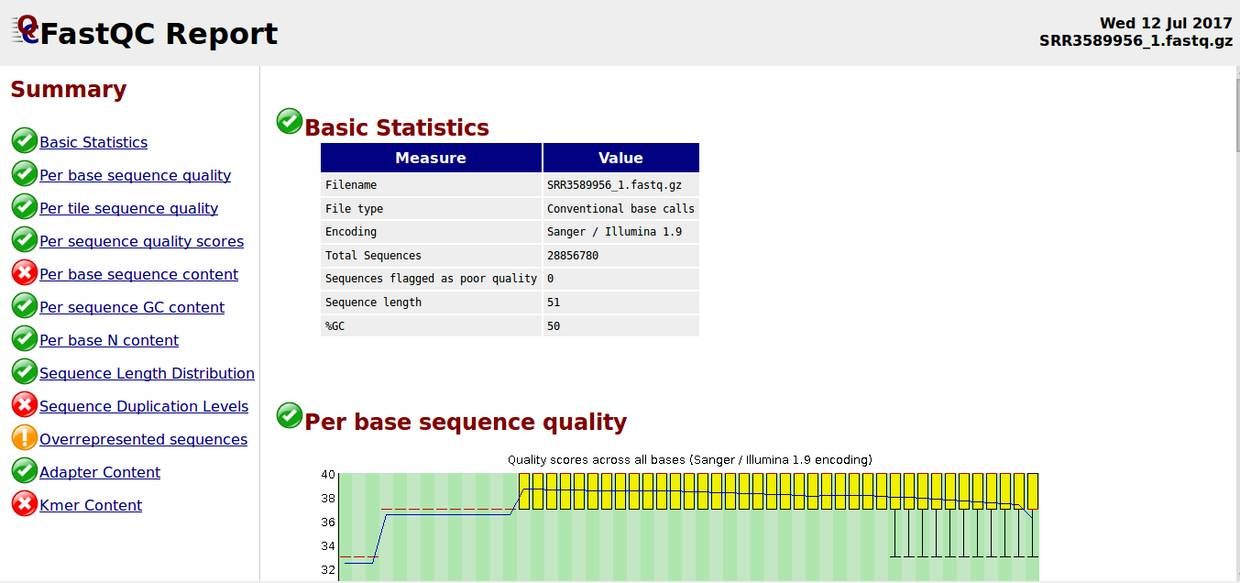
绿色表示“通过”,红色表示“未通过”,黄色表示“警告(不太好)”
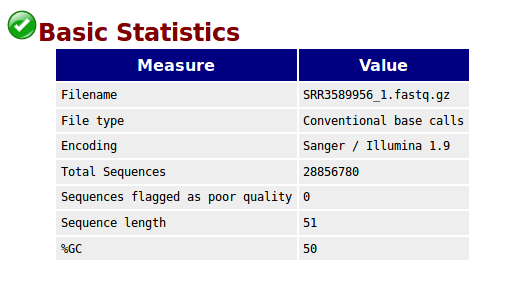
Basic Statistics,基本的数据统计包括文件名,文件类型,编码形式,总的序列数,质量差的序列,序列平均长度,GC含量。
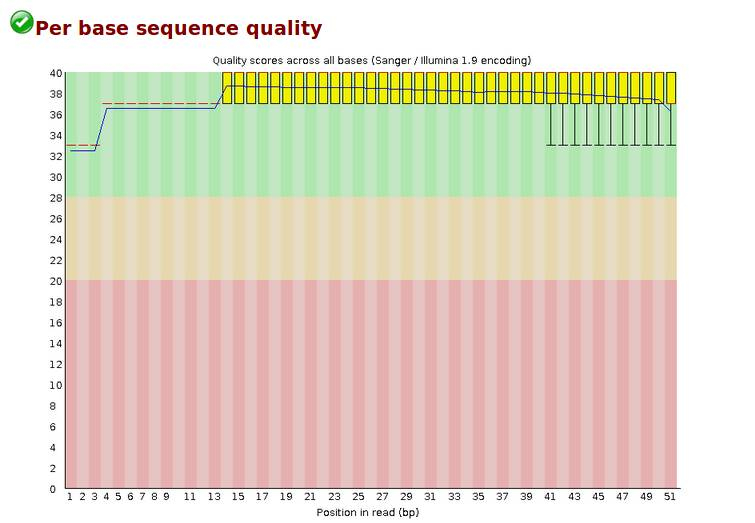
Per base sequence quality,各位置碱基质量,每个read各位置碱基的测序质量。
横轴碱基的位置,纵轴是质量分数,Quality score=-10log10p(p代表错误率),所以当质量分数为40的时候,p就是0.0001,质量算高了。
红色线代表中位数,蓝色代表平均数,黄色是25%-75%区间,触须是10%-90%区间(黄色和触须我不是特别明白)。若任一位置的下四分位数低于10或者中位数低于25,出现“警告”;若任一位置的下四分位数低于5或者中位数低于20,出现“失败,Fail”。
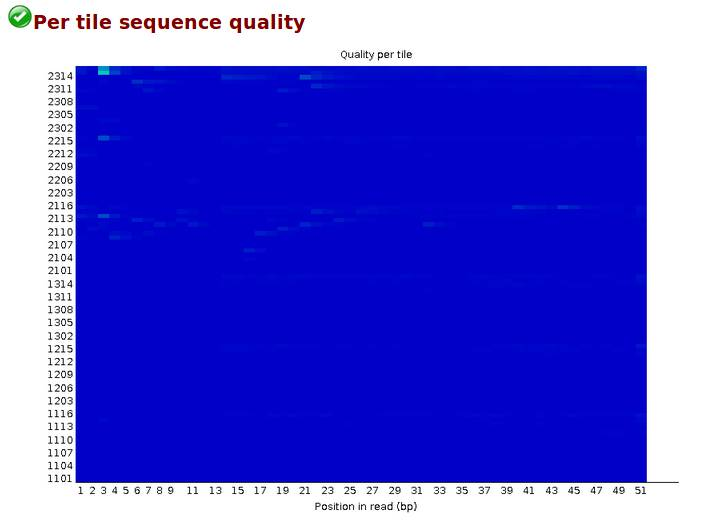
Per tile sequence quality,偏离度,检查reads中每一个碱基位置在不同的测序小孔之间的偏离度,蓝色代表偏离度小,质量好,越红代表偏离度越大,质量越差。
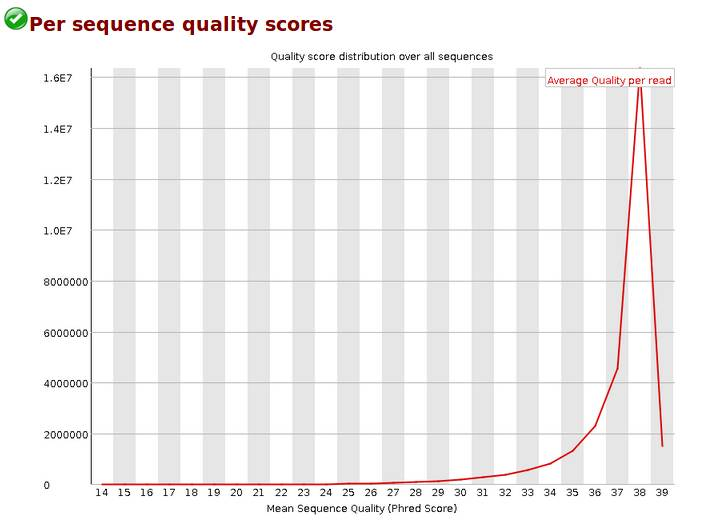
Per sequence quality scores,reads质量的分布,当峰值小于27时,警告;当峰值小于20时,fail。我的报告峰值在38。
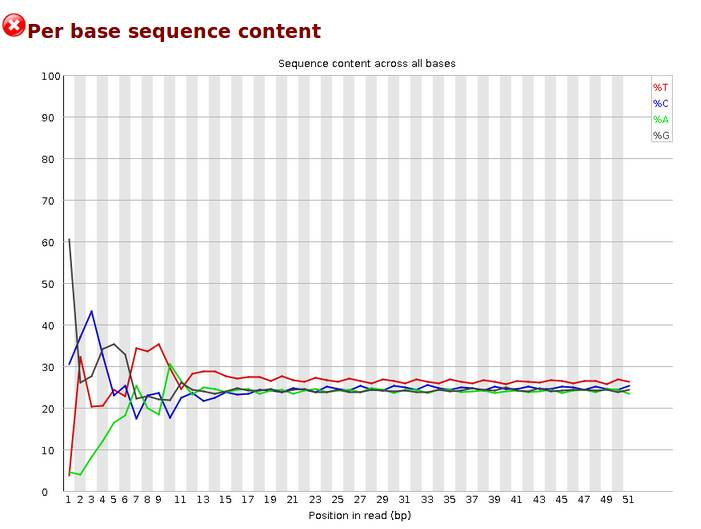
Per base sequence content,碱基分布,对所有reads的每一个位置,统计ATCG四种碱基的分布,横轴为位置,纵轴为碱基含量,正常情况下每个位置每种碱基出现的概率是相近的,四条线应该平行且相近。当部分位置碱基的比例出现bias时,即四条线在某些位置纷乱交织,往往提示我们有overrepresented sequence的污染。本结果前10个位置,每种碱基频率有明显的差别,说明有污染。当任一位置的A/T比例与G/C比例相差超过10%,报'WARN';当任一位置的A/T比例与G/C比例相差超过20%,报'FAIL'。
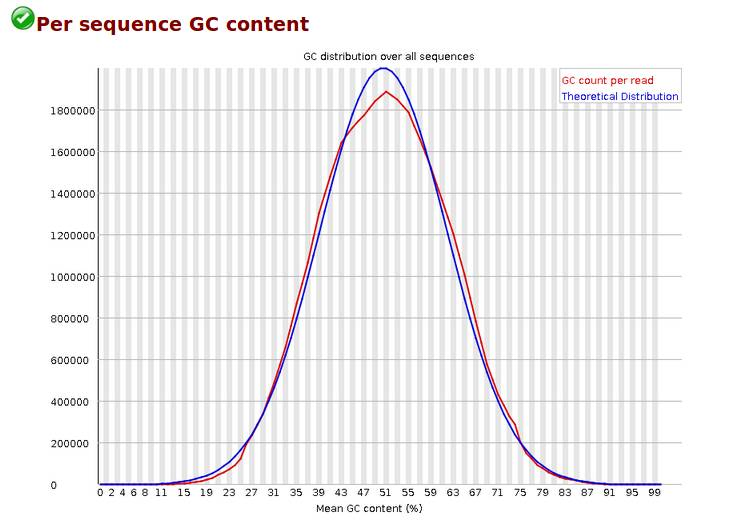
Per Sequence GC Content,reads 平均GC含量分布,统计reads的平均GC含量的分布。红线是实际情况,蓝线是理论分布(正态分布,均值不一定在50%,而是由平均GC含量推断的)。 曲线形状的偏差往往是由于文库的污染或是部分reads构成的子集有偏差(overrepresented reads)。形状接近正态但偏离理论分布的情况提示我们可能有系统偏差。偏离理论分布的reads超过15%时,报'WARN';偏离理论分布的reads超过30%时,报'FAIL'。
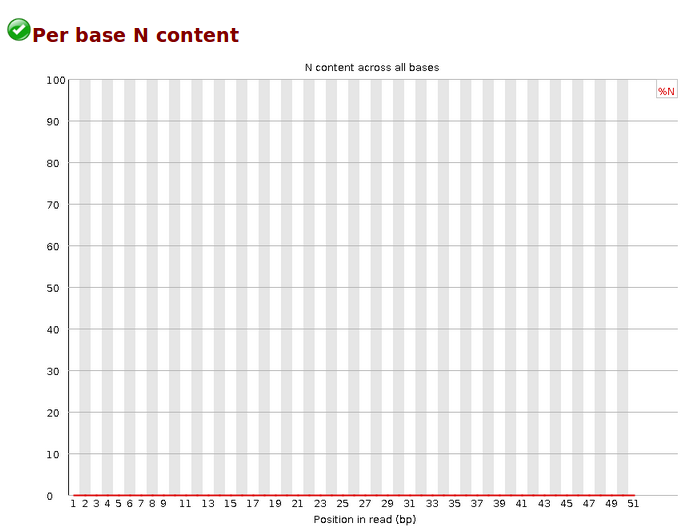
Per base N content,各位置N的reads比率,当测序仪器不能辨别某条reads的某个位置到底是什么碱基时,就会产生“N”,统计N的比率。正常情况下,N值非常小。当任意位置的N的比例超过5%,报'WARN';当任意位置的N的比例超过20%,报'FAIL'。
Sequence Length Distribution,reads长度分布,当reads长度不一致时报'WARN';当有长度为0的read时报“FAIL”。 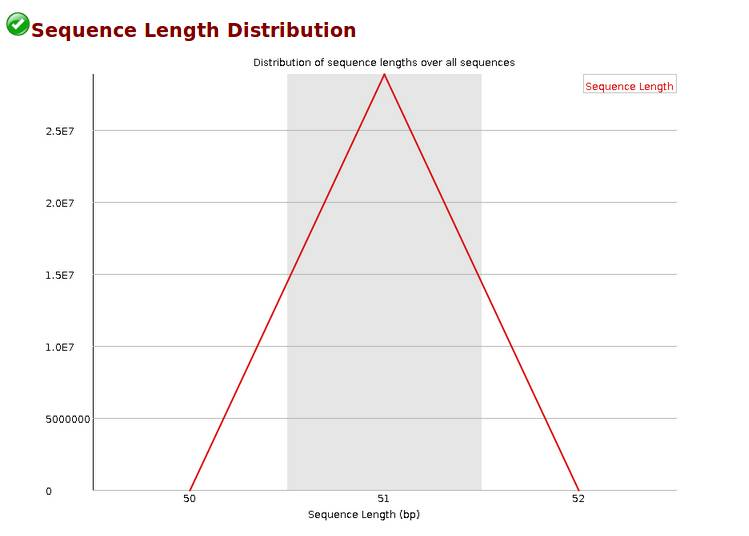
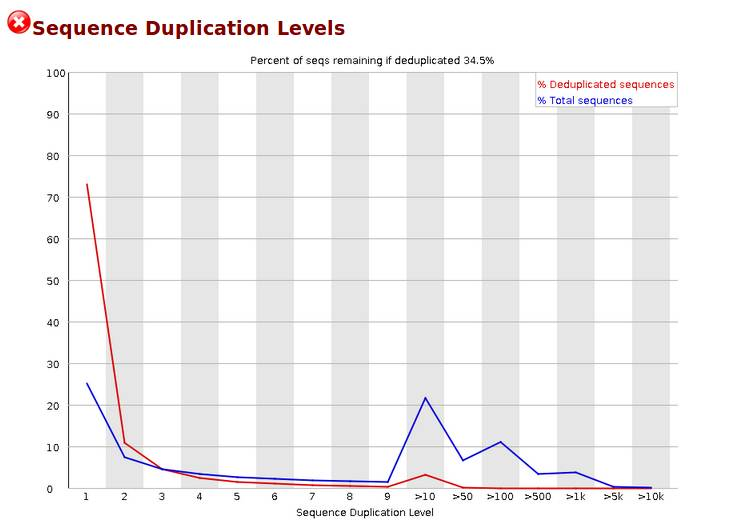
Sequence Duplication Levels,统计不同拷贝数的reads的频率。测序深度越高,越容易产生一定程度的duplication,这是正常的现象,但如果duplication的程度很高,就提示我们可能有bias的存在。横坐标是duplication的次数,纵坐标是duplicated reads的数目,以unique reads的总数作为100%。下图中,大于10个重复的reads占总序列的20%以上,其他依次类推。当非unique的reads占总数的比例大于20%时,报'WARN';当非unique的reads占总数的比例大于50%时,报'FAIL“。
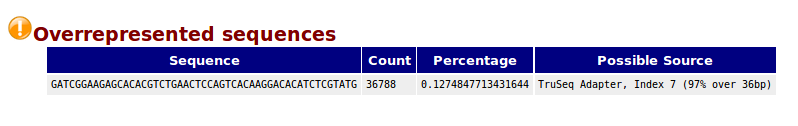
Overrepresented sequences,一条序列的重复数,因为一个转录组中有非常多的转录本,一条序列再怎么多也不太会占整个转录组的一小部分(比如1%),如果出现这种情况,不是这种转录本巨量表达,就是样品被污染。这个模块列出来大于全部转录组1%的reads序列,但是因为用的是前200,000条,所以其实参考意义不大,完全可以忽略。
Adapter content,接头含量
Kmer content
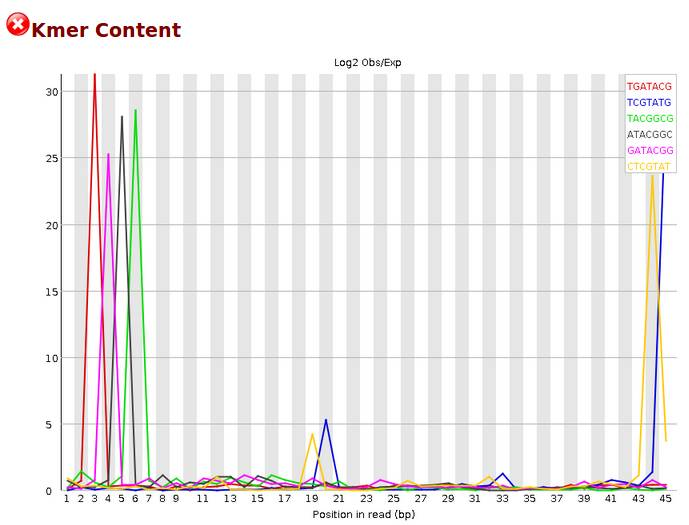
一般而言RNA-Seq数据在sequence deplication levels 未通过是比较正常的。毕竟一个基因会大量表达,会测到很多遍
用FastQC检查二代测序原始数据的质量 | Public Library of Bioinformatics
https://www.plob.org/article/5987.html
用cutadapt软件来对双端测序数据取出接头
对数据进行QC的3个大方向:quality trimming, adapter removal, contaminant filtering.
1、用fastqc软件对数据进行检测,看有什么接头

接头查询地点:
在github可以查到:https://github.com/csf-ngs/fastqc/blob/master/Contaminants/contaminant_list.txt
或者:Download common Illumina adapters from https://github.com/vsbuffalo/scythe/blob/master/illumina_adapters.fa
TruSeq Universal Adapter: AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCT
Illumina Small RNA 3p Adapter: 1 ATCTCGTATGCCGTCTTCTGCTTG
参考资料
HOPTOP转录组入门(三):你懂质量控制吗?-转录组-生信技能树
http://www.biotrainee.com/thread-1831-1-1.html
转录组入门3-测序数据质量检查 | 分享自为知笔记
http://fbb84b26.wiz03.com/share/s/3XK4IC0cm4CL22pU-r1HPcQQ2irG2836uQYm2iZAyh1Zwf3_
PANDA姐的转录组入门(3):了解fastq测序数据
http://mp.weixin.qq.com/s/1eaNhzj1R5pQgn7uy8Y7OA
(3)转录组之数据质控-转录组-生信技能树
http://www.biotrainee.com/thread-1913-1-1.html
转录组(3):了解fastq测序数据 - 简书
https://www.jianshu.com/p/1e609d106377
【转录组入门】3:了解fastq测序数据的更多相关文章
- 弗雷塞斯 从生物学到生物信息学到机器学习 转录组入门(3):了解fastq测序数据
sra文件转换为fastq格式 1 fastq-dump -h --split-3 也就是说如果SRA文件中只有一个文件,那么这个参数就会被忽略.如果原文件中有两个文件,那么它就会把成对的文件按*_1 ...
- 转录组入门(3):了解fastq测序数据
sra文件转换为fastq格式 fastq-dump -h --split-3 也就是说如果SRA文件中只有一个文件,那么这个参数就会被忽略.如果原文件中有两个文件,那么它就会把成对的文件按*_1.f ...
- 单细胞转录组测序数据的可变剪接(alternative splicing)分析方法总结
可变剪接(alternative splicing),在真核生物中是一种非常基本的生物学事件.即基因转录后,先产生初始RNA或称作RNA前体,然后再通过可变剪接方式,选择性的把不同的外显子进行重连,从 ...
- fastx_toolkit去除测序数据中的接头和低质量的reads
高通量测序数据下机后得到了fastq的raw_data,通常测序公司在将数据返还给客户之前会做"clean"处理,即得到clean_data.然而,这些clean_data是否真的 ...
- 测序数据质控-FastQC
通常我们下机得到的数据是raw reads,但是公司通常会质控一份给我们,所以到很多人手上就是clean data了.我们再次使用fastqc来进行测序数据质量查看以及结果分析. fastqc的操作: ...
- Next generation sequencing (NGS)二代测序数据预处理与分析
二代测序原理: 1.DNA待测文库构建. 超声波把DNA打断成小片段,一般200--500bp,两端加上不同的接头2.Flowcell.一个flowcell,8个channel,很多接头3.桥式PCR ...
- 基于单细胞测序数据构建细胞状态转换轨迹(cell trajectory)方法总结
细胞状态转换轨迹构建示意图(Trapnell et al. Nature Biotechnology, 2014) 在各种生物系统中,细胞都会展现出一系列的不同状态(如基因表达的动态变化等),这些状态 ...
- Sqoop2入门之导入关系型数据库数据到HDFS上(sqoop2-1.99.4版本)
sqoop2-1.99.4和sqoop2-1.99.3版本操作略有不同:新版本中使用link代替了老版本的connection,其他使用类似. sqoop2-1.99.4环境搭建参见:Sqoop2环境 ...
- ASP.NET MVC 入门8、ModelState与数据验证
原帖地址:http://www.cnblogs.com/QLeelulu/archive/2008/10/08/1305962.html ViewData有一个ModelState的属性,这是一个类型 ...
随机推荐
- python 读取默认配置文件和用户配置文件 configs
优先从configs_default中读取配置,但是configs_override中的配置可以override它 import configs_default #import configs_ove ...
- 在进行多次scanf时,printf输出错误
随便一处代码,经过改正后,输出正确的 ''' #include <stdio.h> int main(){ int T; scanf("%d",&T ...
- springboot+mybatis-puls利用swagger构建api文档
项目开发常采用前后端分离的方式.前后端通过API进行交互,在Swagger UI中,前后端人员能够直观预览并且测试API,方便前后端人员同步开发. 在SpringBoot中集成swagger,步骤如下 ...
- tornado上帝视角第一次建立WEB服务器
import tornado.ioloop import tornado.web 该视角建立在SOCKET服务端和客户端的基础上. class MainHandler(tornado.web.Requ ...
- CMDB-(paramiko模块 -- 实现ssh连接)
import paramiko # 实现ssh功能的模块 ssh = paramiko.SSHClient() # 实例化对象 ssh.set_missing_host_key_policy(para ...
- [Python]_ELVE_centos7安装Python3.7.1与Python2共存
注:该博客转载至PengYunjing博客,加以改动. #0x01 安装依赖环境 yum -y install zlib-devel bzip2-devel openssl-devel ncurses ...
- visual studio 2017 中默认无法开发 Android 8.0 及以上系统的解决方案
一般默认比较旧有两个原因,系统版本过旧,Visual Studio 版本过旧. 第一步,将windows 更新到最新版,必须是windows 10 并且更新到最新. 第二步,将visual studi ...
- KiCad EDA 原理图库的最佳实践
KiCad EDA 原理图库的最佳实践 由于有 Alias 别名元件,可以不用一个每一个元件都有一个元件. 对每种元件类型建议一个元件库. 因为 Value 和 元件名是一样的,所以元件名要尽可能的简 ...
- 配置django项目总结
1.在django项目应用文件夹中的models.py文件中导入(1)from django.db import models(2)建立需要的映射的类名和属性类型也就是数据库中的表名和字段名 2.在s ...
- 18.25 JLink调试程序步骤
S3C2440开发板启动时候选择NandFlash启动,然后输入如下命令: r /*复位cpu*/ h ...