全基因组关联分析(Genome-Wide Association Study,GWAS)流程
全基因组关联分析流程:
一、准备plink文件
1、准备PED文件
PED文件至少有六列,内容如下:
Family ID
Individual ID
Paternal ID
Maternal ID
Sex (1=male; 2=female; other=unknown)
Phenotype(-9 missing 0 missing 1 unaffected 2 affected)
genotype( 1,2,3,4 or A,C,G,T missing 0)
PED文件是空格(空格或制表符)分隔的文件。
PED文件长这个样:

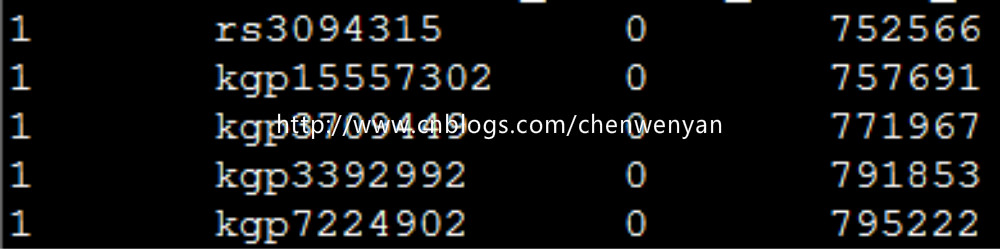
2、准备MAP文件
MAP文件有四列,四列内容如下:
chromosome (1-22, X, Y or 0 if unplaced)
rs# or snp identifier
Genetic distance (morgans)
Base-pair position (bp units)
MAP文件长这个样:
3、生成bed、fam、bim、文件
输入命令
plink --file mydata --out mydata --make-bed
注:plink指的是plink软件,如果软件安装在某个指定的路径的话,前面还要加上路径,比如安装在路径为/your/pathway/的文件夹下,则命令应该为“/your/pathway/plink --file mydata --out mydata --make-bed”
mydata指的是1和2生成的PED和MAP文件名,不需要写.ped和.map后缀
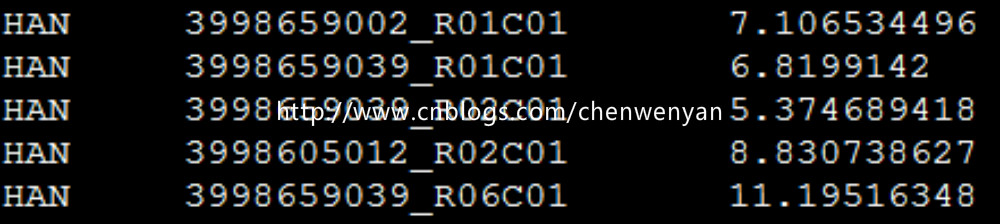
二、准备表型文件(Alternate phenotype files)
一般表型文件为txt格式,表型文件有三列,分别为:
Family ID
Individual ID
Phenotype
假如有多种表型,第一列和第二列还是Family ID、Individual ID,第三列及以后的每列都是表型,例如以下:
Family ID
Individual ID
Phenotype A
Phenotype B
Phenotype C
Phenotype D
Phenotype E
……
表型文件长这样:
缺失值的处理:缺失值的表型用-9表示;
case和control的处理:通常情况下,1表示control,2表示case,0表示缺失,但如果你加上--1的参数,则0表示control,1表示case。
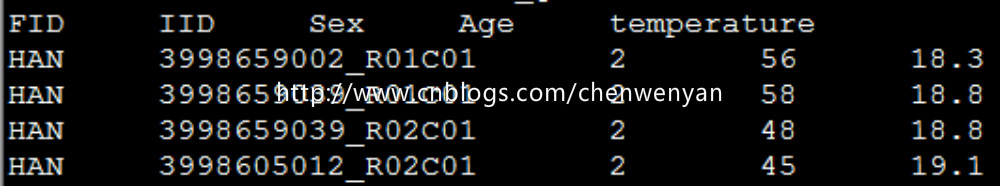
三、准备协变量文件(Covariate files)
协变量文件同表型文件类似,第一列和第二列是Family ID、Individual ID,第三列及以后的每列都是协变量
Family ID
Individual ID
Covariate A
Covariate B
Covariate C
Covariate D
Covariate E
……
协变量文件长这个样(这里有三个协变量,分别为Sex,Age,temperature):
四、plink进行表型和基因型以及协变量的关联分析
命令如下:
plink --bfile mydata --linear --pheno pheno.txt --mpheno 1 --covar covar.txt --covar-number 1,2,3 --out mydata –noweb
生成的文件为mydata.assoc.linear
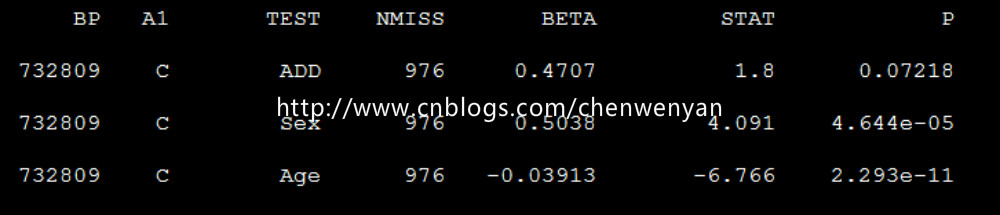
注:“mydata”mydata文件不需要后缀,“--mpheno 1”指的是表型文件的第三列(即第一个表型)
“--covar-number 1,2,3”指的是协变量文件的第三列、第四列、第五列(即第一个、第二个、第三个协变量)
“--linear”指的是用的连续型线性回归,如果表型为二项式(即0、1)类型,则用“--logistic”
五、画曼哈顿图
安装R语言的CpGassoc包,其中的manhattan(),即可画曼哈顿图,或者参照本文R语言画全基因组关联分析中的曼哈顿图(manhattan plot)
全基因组关联分析(Genome-Wide Association Study,GWAS)流程的更多相关文章
- GWAS 全基因组关联分析 | summary statistic 概括统计 | meta-analysis 综合分析
有很多概念需要明确区分: 人有23对染色体,其中22对常染色体autosome,另外一对为性染色体sex chromosome,XX为女,XY为男. 染色体区带命名:在标示一特定的带时需要包括4项:① ...
- 【GWAS文献解读】疟原虫青蒿素抗药性的全基因组关联分析
英文名:Genetic architecture of artemisinin-resistant Plasmodium falciparum 中文名:疟原虫青蒿素抗药性的全基因组关联分析 期刊:Na ...
- 全基因组关联分析(GWAS)的计算原理
前言 关于全基因组关联分析(GWAS)原理的资料,网上有很多. 这也是我写了这么多GWAS的软件教程,却从来没有写过GWAS计算原理的原因. 恰巧之前微博上某位小可爱提问能否写一下GWAS的计算原理. ...
- GWAS | 全基因组关联分析 | Linkage disequilibrium (LD)连锁不平衡 | 曼哈顿图 Manhattan_plot | QQ_plot | haplotype phasing
现在GWAS已经属于比较古老的技术了,主要是碰到严重的瓶颈了,单纯的snp与表现的关联已经不够,需要具体的生物学解释,这些snp是如何具体导致疾病的发生的. 而且,大多数病找到的都不是个别显著的snp ...
- 全基因组关联分析(GWAS):为何我的QQ图那么飘
前段时间有位小可爱问我,为什么她的QQ图特别飘,如果你不理解怎样算飘,请看下图: 理想的QQ图应该是这样的: 我当时的第一反应是:1)群体分层造成的:2)表型分布有问题.因此让她检查一下数据的群体分层 ...
- 一行命令学会全基因组关联分析(GWAS)的meta分析
为什么需要做meta分析 群体分层是GWAS研究中一个比较常见的假阳性来源. 也就是说,如果数据存在群体分层,却不加以控制,那么很容易得到一堆假阳性位点. 当群体出现分层时,常规手段就是将分层的群体独 ...
- 全基因组关联分析(GWAS)扫不出信号怎么办(文献解读)
假如你的GWAS结果出现如下图的时候,怎么办呢?GWAS没有如预期般的扫出完美的显著信号,也就没法继续发挥后续研究的套路了. 最近,nature发表了一篇文献“Common genetic varia ...
- R语言画全基因组关联分析中的曼哈顿图(manhattan plot)
1.在linux中安装好R 2.准备好画曼哈顿图的R脚本即manhattan.r,manhattan.r内容如下: #!/usr/bin/Rscript #example : Rscript plot ...
- 全基因组关联分析学习资料(GWAS tutorial)
前言 很多人问我有没有关于全基因组关联分析(GWAS)原理的书籍或者文章推荐. 其实我个人觉得,做这个分析,先从跑流程开始,再去看原理. 为什么这么说呢,因为对于初学者来说,跑流程就像一个大黑洞,学习 ...
随机推荐
- Linux学习二:Makefile基础
文首感谢http://www.chinaunix.net 作者:gunguymadman的分享 makefile关系到了整个工程的编译规则.一个工程中的源文件不计数,其按类型.功能.模块分别放在若干个 ...
- 这些年正Android - 母亲
记得小时候,在自己写完一篇作文,完成母亲布置的任务后,就会搬走母亲正在使用的大椅子,面朝门前的马路,就这么憧憬的坐着,听着母亲给小孩打针时,小孩哇哇的哭声,努力的幻想着自己以后能做一个顶天立地的男子汉 ...
- angularjs中文社区
http://angularjs.cn/ 中文社区 https://angular.cn/features.html 官方文档中文版
- windows7 编译boost1.54
先去下载boost1.54 for windows原码.原来有个安装工具现在已经停止维护了,我试了旧版,已经安装不了. 这是它们的网站:http://www.boostpro.com/download ...
- shellinabox基于web浏览器的终端模拟器
1. Shellinabox介绍 Shellinabox 是一个利用 Ajax 技术构建的基于 Web 浏览器的远程终端模拟器,也就是说安装了该软件之后,服务器端不需要开启 ssh服务,通过 Web ...
- shell 脚本 exit 1 报错:numeric argument required问题解决
原因是在window环境编辑会有特殊字符,解决办法:sed -i'' "s/\r//" file_name
- 线程+IO流
第十八天知识点总结 线程的停止: 1.停止一个线程,一般是通过一个变量来控制. 2.如果需要停止一个处于一个等待状态的线程,那么需要配合interrupt方法来完成 守护线程(后台线程):在一个进程中 ...
- GPL 和BSD和Apache
开源许可证GPL.BSD.MIT.Mozilla.Apache和LGPL的区别<ignore_js_op> 以下是上述协议的简单介绍:BSD开源协议BSD开源协议是一个给于使用者很大自由的 ...
- [整理]FPGA学习资料汇总
01.特权同学倾情奉献海量FPGA学习资料 http://pan.baidu.com/s/1pJIb32F
- BeanUtils.populate的作用
它是在org.apache.commons.beanutils.BeanUtils包中的一个方法. 方法的作用:将一些 key-value 的值(例如 hashmap)映射到 bean 中的属性. ...