使用RSEM进行转录组测序的差异表达分析
仍然是两年前的笔记
1. prepare-reference
如果用RSEM对比对后的bam进行转录本定量,则在比对过程中要确保比对用到的索引是由rsem-prepare-reference产生的。
~/software/rsem/rsem-prepare-reference \
--transcript-to-gene-map ~/project/RNA-seq/ref_cds/gene_transcript.txt \ #作用是在后面的定量结果文件中,添加gene名称, 转录本名称两列,该文件每一行都是gene_id\ttranscript_id的形式,eg: cluster_11236 cluster_11236.1
--bowtie2 \ #RSEM可调用bowtie, bowtie2, STAR三种比对工具;这里选用bowtie2
~/project/RNA-seq/ref_cds/HC_cds_and_8sample_clustercds.fa \
~/project/RNA-seq/ref_cds/cds.byrsem

可以看到,单纯用bowtie2建的索引和rsem调用bowtie2建的索引是不一样的。
2. calculate-expression
用法分为两类,分别是从fa/fq得到表达矩阵,和从sam/bam得到表达矩阵(仍然要求是比对到rsem-prepare-reference生成的索引)。以单端的fq数据为例。
rsem-calculate-expression [options] upstream_read_file(s) reference_name sample_name
rsem-calculate-expression [options] --paired-end upstream_read_file(s) downstream_read_file(s) reference_name sample_name
rsem-calculate-expression [options] --sam/--bam [--paired-end] input reference_name sample_name
cat ~/project/RNA-seq/dir.txt | while read id
do
~/software/rsem/rsem-calculate-expression -p 8 --bowtie2 \
~/project/data/RNA-seq/${id}.fastq.gz \
~/project/RNA-seq/ref_cds/cds.byrsem \
--samtools-sort-mem 2G --fragment-length-mean 50 \ # 单端数据建议使用--fragment-length-mean和--fragment-length-sd
~/project/RNA-seq/map/${id}.rsem
done
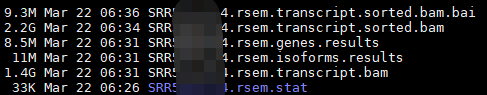
完成之后得到这些文件,其中,rsem.genes.results和rsem.isoforms.results分别表示gene水平和转录本水平的定量结果。每一列含义:
less rsem.genes.results|head -n 1
gene_id transcript_id(s) length effective_length expected_count TPM FPKM
less rsem.isoforms.results|head -n 1
transcript_id gene_id length effective_length expected_count TPM FPKM IsoPct
后面用EBseq检验差异基因/转录本时,会使用到这两个文件。
3. Differential Expression Analysis using EBSeq
下面以gene水平差异分析为例。
3.1 generate-data-matrix
这一步提取上一步得到的每个样本定量结果文件中的expected_count列,组成数据矩阵。
~/software/rsem/rsem-generate-data-matrix \
SRR1.rsem.genes.results SRR2.rsem.genes.results \
SRR3.rsem.genes.results SRR4.rsem.genes.results \
SRR5.rsem.genes.results SRR6.rsem.genes.results \
SRR7.rsem.genes.results SRR8.rsem.genes.results \
> ~/project/RNA-seq/count/GeneMat.txt
3.2 run-ebseq
调用EBseq进行检验
~/software/rsem/rsem-run-ebseq \
GeneMat.txt 2,2,2,2 GeneMat.results #2,2,2,2表示4个condition, 每个condition有两个重复;顺序要和3.1中输入文件表示的condition的顺序一致
#会得到三个文件
GeneMat.results.condmeans GeneMat.results GeneMat.results.pattern
#GeneMat.results.pattern
"C1" "C2" "C3" "C4"
"Pattern1" 1 1 1 1
"Pattern2" 1 1 1 2
"Pattern3" 1 1 2 1
"Pattern4" 1 1 2 2
"Pattern5" 1 2 1 1
"Pattern6" 1 2 1 2
"Pattern7" 1 2 2 1
"Pattern8" 1 2 2 2
"Pattern9" 1 1 2 3
"Pattern10" 1 2 1 3
"Pattern11" 1 2 2 3
"Pattern12" 1 2 3 1
"Pattern13" 1 2 3 2
"Pattern14" 1 2 3 3
"Pattern15" 1 2 3 4
#以Pattern14为例,1 2 3 3表示某基因表达:C1与C2不同,C3与C4相同
#四种condition如果有基因表达存在差异,就这些情况了
#GeneMat.results
#第一列是各个基因名称,接着15列是该基因符合该种Parttern的概率
#"MAP"为该基因最可能的模式;"PPDE":posterior probability of being differentially expressed,越大越好
"Pattern1" "Pattern2" "Pattern3" "Pattern4" "Pattern5" "Pattern6" "Pattern7" "Pattern8" "Pattern9" "Pattern10" "Pattern11" "Pattern12" "Pattern13" "Pattern14" "Pattern15" "MAP" "PPDE"
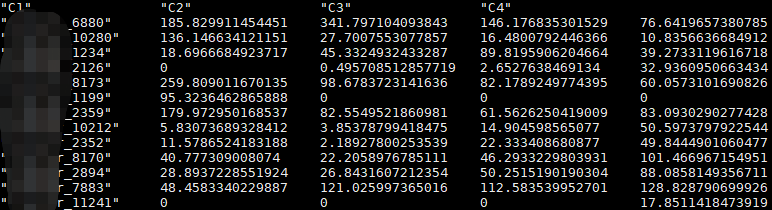
#GeneMat.results.condmeans
#为每个样本合并重复之后的定量结果,如下图,这个结果可以用来控制fold change
3.3 control_fdr
控制FDR(错误发现率)来挑选差异基因
~/software/rsem/rsem-control-fdr \
GeneMat.results 0.05 GeneMat.de.txt
将GeneMat.results文件中,PPDE大于0.95的记录提取出来
因水平有限,有错误的地方,欢迎批评指正!
使用RSEM进行转录组测序的差异表达分析的更多相关文章
- 转录组差异表达分析工具Ballgown
Ballgown是分析转录组差异表达的R包. 软件安装: 运行R, source(“http://bioconductor.org/biocLite.R”) biocLite(“ballgown”) ...
- 单细胞转录组测序数据的可变剪接(alternative splicing)分析方法总结
可变剪接(alternative splicing),在真核生物中是一种非常基本的生物学事件.即基因转录后,先产生初始RNA或称作RNA前体,然后再通过可变剪接方式,选择性的把不同的外显子进行重连,从 ...
- 差异表达分析之FDR
差异表达分析之FDR 随着测序成本的不断降低,转录组测序分析已逐渐成为一种很常用的分析手段.但对于转录组分析当中的一些概念,很多人还不是很清楚.今天,小编就来谈谈在转录组分析中,经常会遇到的一个概念F ...
- Differential expression analysis for paired RNA-seq data 成对RNA-seq数据的差异表达分析
Differential expression analysis for paired RNA-seq data 抽象背景:RNA-Seq技术通过产生序列读数并在不同生物条件下计数其频率来测量转录本丰 ...
- RNA-Seq differential expression analysis: An extended review and a software tool RNA-Seq差异表达分析: 扩展评论和软件工具
RNA-Seq differential expression analysis: An extended review and a software tool RNA-Seq差异表达分析: 扩展 ...
- 表达谱(DGE)测序与转录组测序的差别
DGE-seq和普通的transcriptomic profiling相比较有什么不同,有什么特点? DGE就是用酶将mRNA切断,只使用靠近poly A的一小段RNA去测序. #1 由于不是测定mR ...
- 单细胞转录组测序技术(scRNA-seq)及细胞分离技术分类汇总
单细胞测序流程(http://learn.gencore.bio.nyu.edu) 在过去的十多年里,高通量测序技术被广泛应用于生物和医学的各种领域,极大促进了相关的研究和应用.其中转录组测序(RNA ...
- 转录组测序(RNA-seq)技术
转录组是某个物种或者特定细胞类型产生的所有转录本的集合.转录组研究能够从整体水 平研究基因功能以及基因结构,揭示特定生物学过程以及疾病发生过程中的分子机理,已广泛应 用于基础研究.临床诊断和药 ...
- 转录组分析综述A survey of best practices for RNA-seq data analysis
转录组分析综述 转录组 文献解读 Trinity cufflinks 转录组研究综述文章解读 今天介绍下小编最近阅读的关于RNA-seq分析的文章,文章发在Genome Biology 上的A sur ...
随机推荐
- Linux——软件安装
Linux--软件安装 一.gcc 二.make 三.rpm 四.yum 一.gcc gcc是Linux上面最标准的C语言的编译程序,用来源代码的编译链接. gcc -c hello.c 编译产生目标 ...
- MySql命令,吐血整理的Mysql,实话,真的吐血
MySql命令,吐血整理的Mysql,实话,真的吐血 1.基本操作 2.数据库操作 3.表的操作 4.数据操作 5.字符集编码 6.数据类型(列类型) 7.列属性(列约束) 8.建表规范 9.SELE ...
- Gitlab + DRBD HA
部署简介: 为了gitlab有容灾的能力,所以部署一个HA的小集群,用到的软件有 gitlab 和brbd,目前现有环境为 master节点 系统版本:CentOS release 6.5 (Fina ...
- hadoop(集群)完全分布式环境搭建
一,环境 主节点一台: ubuntu desktop 16.04 zhoujun 172.16.12.1 从节点(slave)两台:ubuntu server 16.04 hadoop2 ...
- 跟着Vimtutor学习Vim
跟着Vimtutor学习Vim Lesson 1 1.1 移动光标 在Vim中移动光标,分别使用h.j.k.l键代表左.下.上.右方向. 1.2 退出VIM :q! <ENTER> 退出V ...
- php之魔术方法 __set(),__get(),__isset(),__unset()
__set()与__get() 当一个类里面,属性被设置为私有属性时,这个属性是不能在外部被访问的.那么当我们又想在外部访问时该怎么办呢,我们可以用方法来实现.举例如下: 1 class Test 2 ...
- 设计模式(六)——建造者模式(源码StringBuilder分析)
建造者模式 1 盖房项目需求 1) 需要建房子:这一过程为打桩.砌墙.封顶 2) 房子有各种各样的,比如普通房,高楼,别墅,各种房子的过程虽然一样,但是要求不要相同的. 3) 请编写程序,完成需求. ...
- P1908 逆序对——树状数组&离散化&快读快写の学习
题目简述: 对于给定的一段正整数序列,逆序对就是序列中 a_i>a_jai>aj 且 i<ji<j 的有序对. 输出序列中逆序对的数目. 知识补充: 树状数组: 这东西就是 ...
- C#遇见C++的江湖 不行!得不到就干掉
C# VS C++ C#代码遇见了非托管dll如何处理 问题:托管与非托管,兼容? 方法一:DllImport 托管调试助手 "PInvokeStackImbalance" Me ...
- Shiro权限项目
目录 环境配置 spring容器 springmvc freemarker mybatis shiro 工具类 TokenManager.java Result.java 功能实现 登录 注册 个人中 ...