Sam format
reference:https://davetang.org/wiki/tiki-index.php?page=SAM
@SQ SN:contig1 LN:9401 (序列ID及长度)
参考序列名,这些参考序列决定了比对结果sort的顺序,SN是参考序列名;LN是参考序列长度;每个参考序列为一行。
@PG ID:bowtie2 PN:bowtie2 VN:2.0.0-beta7 (比对所使用的软件及版本)
这里的ID是bwa,PN是bwa,VN是0.7.12-r1039版本。CL可以认为是运行程序
K00133::H5LL5BBXX:::: * * * 0A NCCATCCATAATTTATAGGGAAGTGTTAAAATGCGAATAATATCCTTTTCTTTTCCTGGGATTCGTTCAACATTTCCATTTCCATTTCCCTCCGAGTT GATTTATGGGAATTCTCAGCAGCCTTGTCCATAATTCATGAACAATCCCTG A#AFFAFFJJJJJJJJJJJJJJ7FJJJJAJJJJJJJJJJFJJ JJJJJJJJJJJJJAJJJJJJJJJFJAJJJJJFJJFJJJJJJJJJJJJFFFJJJFA7AA7FFJAJ<AJ<AFF-FFFJFJFJJJF<AFFAFAFJJFAJJJ FA<<<AFJJA AS:i: XS:i:
K00133::H5LL5BBXX:::: * * * 0N AAGGAGAAGTTCCCATAGCAACAGCACCATTTTTTCCAGGGAAAAGGAGGAAAGAATCAAGGATGCGAAAGAGGGAATCAAAGCAGCTTAAGGGATAT AAAAAAAACAGGGATTGTTCATGAATTATGGACAAGGCAGCTGAGAATTCC #AAFFJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJA-<- 7A<7AJJF<AJJJJJF7JFAJF7AJJFJJJJJJJFF-AFJFJJJJJ--FFJJJJJJAAJFF<AJJJJJF777-----<-7F7-<--<<<<<7F<--) --A7FF--< AS:i: XS:i:
K00133::H5LL5BBXX:::: * * * 0T NTTTGAGTGCCTTCTAATTTTTTTTCTCCTGTATCTAAATTGCATGAGTATACTCTGCTACATTTAATCTTCAGGTAAAACAAAACTTTCTTTTATCA TATTTAGGAAGTATCACTAATCATGAATTAAAATTAAATGTGTATTGTCAG A#AFAJJJJJJJJJJJJJJJJJJJJJJJJJJFJJJFJJJJJJ FJJJFJJJJJJJJFJFJJJJJJJJJJJJJAJJFJAAJ77-AJFJFJJJJJFJJJJJJJFJJJFJF7AFFJAFFJFFA<FFAFJJAFAJFJJJJJJF-A FFJF77-A-- AS:i: XS:i:
K00133::H5LL5BBXX:::: * * * 0N CTGGCAGGCACAGATGCCCACAGTACACACACTGATATTTATAAAGCACTTCACAAAAAGTCACAAAAAAACCCCCAAAAACGAACCAAACCCAAAAA CCCCACCAAGCAAGCTGACAATACACATTTAATTTTAATTCATGATTAGTG #AAFFJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJFJ JFJJJJJJJJJJJJJJJJJAJJJJJJJJJJJJJJFFJJJJJ<JFJFJFJJFJ<JJJJJ<FJJJJJFJJJJF7F<FJFJ7AFAJJ7FFFAF7F-AAF< JFA<-<-< AS:i: XS:i:

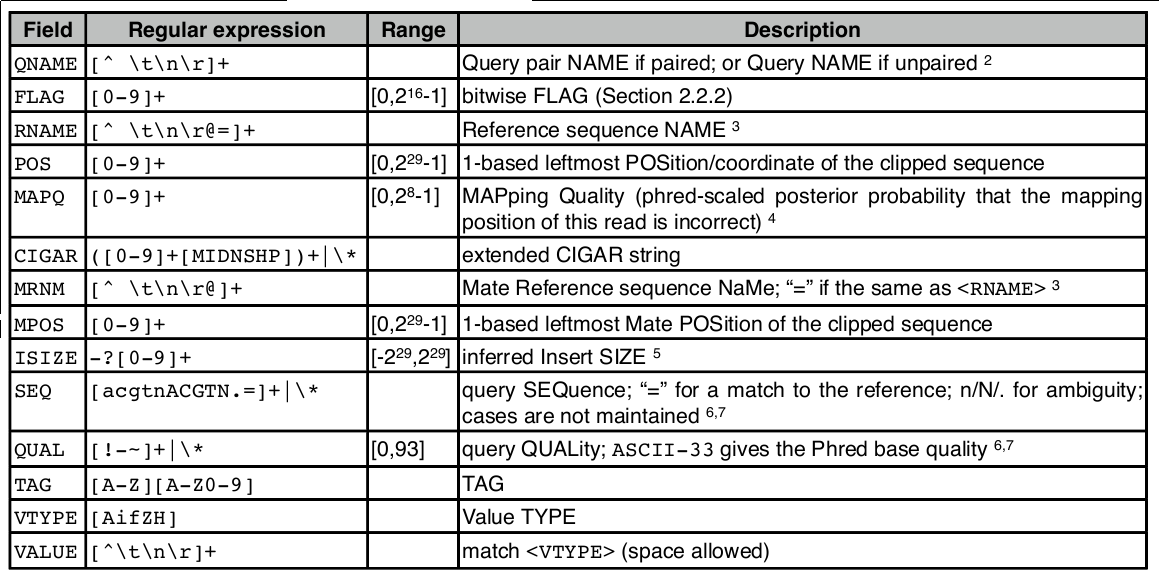
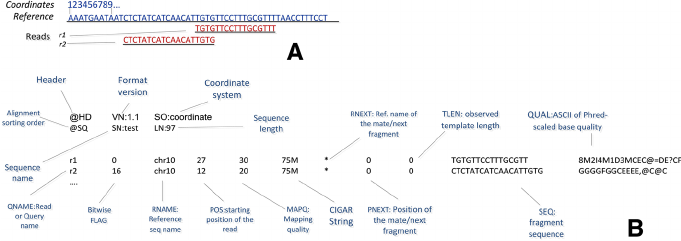
$1:QNAME( 比对的序列名称,即一条测序reads的名称 ):
K00133:143:H5LL5BBXX:8:1101:1194:1103
$2:FLAG(表明比对类型:paring,strand,mate strand等):
可以使用samtools查到flag信息
$ samtools flags
0x4d PAIRED,UNMAP,MUNMAP,READ1 $ samtools flags
0x8d PAIRED,UNMAP,MUNMAP,READ2
$3:RNAME(表示read比对的那条序列的序列名称):
情况一:这条read有比对上的序列,则名称与头部的@SQ相对应,eg:
$ bwa mem -t -M ../.index/t/t1.fa ../.grep/t/o3.gz ../.grep/t/o4.gz >bwa.log |les|awk '{if($3!="*"){print $0}}'
@SQ SN:scaffold332 LN:
@SQ SN:scaffold322 LN:
@SQ SN:scaffold342 LN:
@SQ SN:scaffold191 LN:
@SQ SN:scaffold1157 LN:
@PG ID:bwa PN:bwa VN:0.7.-r1188 CL:bwa mem -t -M ../.index/t/t1.fa ../.grep/t/o3.gz ../.grep/t/o4.gz
K00133::H5LL5BBXX:::: scaffold191 * = CNTCTTTTCAACTTTCCTACCTGGGACACCAAAAGAAGTGAATTTGAAATGCTGGAATACCCATTTCTTTGTAATATACTACCAGGCAATAATTTCTTCACCAGTGTGTGTAAGCCTGTAGCACTGCCTTGTGACCCACGGGCAGGACCC A#AFFJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJFJJJJJJJJJJJJJJJJJJJJJJJJJJJ MC:Z:47S40M63S AS:i: XS:i:
K00133::H5LL5BBXX:::: scaffold191 47S40M63S = NCAAGCTGGGTGGGAGTGTTGATCTGCTGGAGGGCAGGAGGGCTCTGCAGAGGGATCTGGACAGGCTGGATCGATGGGCTGAGGCCAGTTGTATGAGGTTTAATAAGGTGAAATGCTGGGTCCTGCCCGTGGGTCACAAGGCAGTGCTAC #AAFFJJJJJAJJJJJ<JJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJFJJJJJJJJJJFJJJJJJJJJJJJAJJJJFJJFFFJFJJJJJJ-<FJJJJJJJJJJJJFFFJAFJJJJJJJ7FJ<JJJFJJJJJJJJJJJJJ<FF NM:i: MD:Z:26C13 AS:i: XS:i:
情况二:这条read没有比对上的序列,则名称是“*”,且这一行的第四,五,八,九 列是“0”,第六,七列与该列是相同的表示方法
$4:POS(表示read比对到RNAME这条序列的最左边的位置)
noted:
(1)无论该read是正向比对或是反向比对到该序列,比对结果均是正向序列或反向序列最左端的比对位置
(2)如果该read能够完全比对到这条序列,则CIGAR string为M
$5:MAPQ(表示为mapping的质量值)
(1)255:mapping值不可用,
(2)0:通常对于unmapped read
(3)一般结果是这一列的数值是从0到60,且0和60这两个数字出现次数最多
(4)第五列为60表示mapping率最高,
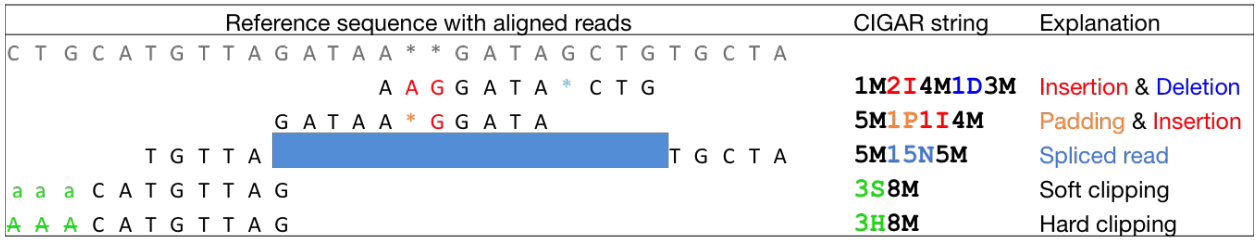
$6:CIGARCIGAR,简要比对信息表达式(Compact Idiosyncratic Gapped Alignment Report),其以参考序列为基础

M:表示read可mapping到第三列的序列上,则read的碱基序列与第三列的序列碱基相同,表示正常的mapping结果,M表示完全匹配,但是无论reads与序列的正确匹配或是错误匹配该位置都显示
I:表示read的碱基序列相对于第三列的RNAME序列,有碱基的插入
D:表示read的碱基序列相对于第三列的RNAME序列,有碱基的删除
N:read 中有gap
P:read 和reference sequence都有gap
S:read 与reference没有mapping上的区域,但是保留了该mismatchs
H:read 与reference没有mapping上的区域,但是没有保留该mismatchs
=:正确匹配到序列上
X:错误匹配到序列上
set ‘*’ if unavailable
而H只出现在一条read的前端或末端,但不会出现在中间,S一般会和H成对出现,当有H出现时,一定会有一个与之对应的S出现
例如:
162M89S
162H89M
149M102S
149H102M
40S211M
20M1D20M211H
S可以单独出现,而H必须有与之对应的S出现时才可能出现,不可在相同第一列的情况下单独出现
N:如果是mRNA-to-genome,N出现的位置代表内含子,其它比对形式出现N时则没有具体解释
M/I/S/=/X:这些数值的加和等于第10列SEQ的长度
如图:

$7:MRNM
情况一:这条reads第二次比对的位置,
在利用bwa mem产生sam文件时,如果该列是“=”而第3列RNAME不是“*”,则表示该reads比对到第3列显示序列名的序列上,而没有比对到其他位置,
在利用bwa aln及bwa sampe比对生成的sam文件,如果和上述情况相同,则第7列为“=”,上述情况均表示该reads只比对到这一个位置
情况二:如果这条reads没有匹配上的序列,第3列RNAME和第7列MRNM都为“*”
情况三:如果这条reads匹配两个序列,则第一个序列的名称出现在第3列,而第二个序列的名称出现在第7列
$8:MPOS:该列表示与该reads对应的mate pair reads的比对位置,
如果这对pair-end reads比对到同一条reference序列上,在sam文件中reads的id出现2次,Read1比对的第4列等于Read2比对的第8列。同样Read1比对的第8列等于Read2比对的第4列。例如:
第1列(Read id)····第4列(Read1比对位置)····第8列(mate-pair reads比对位置)
22699:1759····124057649····124057667
22699:1759····124057667····124057649
相同的reads id一个来自Read1文件,一个来自Read2文件,第4列和第8列是对应的
$ bwa mem -t -M ../.index/t/t1.fa ../.grep/t/o3.gz ../.grep/t/o4.gz >bwa.log |awk '{if($3!="*"){print $0}}'|awk '{print $1"\t"$4"\t"$8}'|tail -
K00133::H5LL5BBXX::::
K00133::H5LL5BBXX::::
$9:TLEN:insert length+mapping base
如果R1端的read和R2端的read能够mapping到同一条Reference序列上(即第三列RNAME相同),则该列的值表示第8列减去第4列加上第6列的M值,即为R1端和R2端相同id的reads其第九列值相同,但该值为一正一负,R1文件的reads和R2文件的reads,相同id的reads要相对来看。在进行该第列值的计算时,如果取第6列的数值,一定要取出现M的值,S或H的值不能取。It is set as 0 for single-segment template or when the information is unavailable.
$ bwa mem -t -M ../.index/t/t1.fa ../.grep/t/o3.gz ../.grep/t/o4.gz >bwa.log |awk '{if($3!="*"){print $0}}'|awk '{print $1"\t"$9}'|tail -
K00133::H5LL5BBXX::::
K00133::H5LL5BBXX::::
$10:SEQ,(序列片段的序列信息,如果不存储此类信息,此处为’*‘,注意CIGAR中M/I/S/=/X对应数字的和要等于序列长度);
$ bwa mem -t -M ../.index/t/t1.fa ../.grep/t/o3.gz ../.grep/t/o4.gz >bwa.log |awk '{if($3!="*"){print $0}}'|awk '{print $1"\t"$10}'|tail -
K00133::H5LL5BBXX:::: CNTCTTTTCAACTTTCCTACCTGGGACACCAAAAGAAGTGAATTTGAAATGCTGGAATACCCATTTCTTTGTAATATACTACCAGGCAATAATTTCTTCACCAGTGTGTGTAAGCCTGTAGCACTGCCTTGTGACCCACGGGCAGGACCC
K00133::H5LL5BBXX:::: NCAAGCTGGGTGGGAGTGTTGATCTGCTGGAGGGCAGGAGGGCTCTGCAGAGGGATCTGGACAGGCTGGATCGATGGGCTGAGGCCAGTTGTATGAGGTTTAATAAGGTGAAATGCTGGGTCCTGCCCGTGGGTCACAAGGCAGTGCTAC
$11:QUAL,(ASCII,read质量的ASCII编码。)
$ bwa mem -t -M ../.index/t/t1.fa ../.grep/t/o3.gz ../.grep/t/o4.gz >bwa.log "$13"\t"$14"\t"$15"\t"$16}'|tail -2
K00133::H5LL5BBXX:::: MC:Z:47S40M63S AS:i: XS:i:
K00133::H5LL5BBXX:::: NM:i: MD:Z:26C13 AS:i: XS:i:
$12之后是自选option
$ bwa mem -t -M ../.index/t/t1.fa ../.grep/t/o3.gz ../.grep/t/o4.gz >bwa.log |awk '{if($3!="*"){print $0}}'|awk '{print $1"\t"$12"\t"$13"\t"$14"\t"$15"\t"$16}'|tail -
K00133::H5LL5BBXX:::: MC:Z:47S40M63S AS:i: XS:i:
K00133::H5LL5BBXX:::: NM:i: MD:Z:26C13 AS:i: XS:i:
Sam format的更多相关文章
- SAMTOOLS使用 SAM BAM文件处理
[怪毛匠子 整理] samtools学习及使用范例,以及官方文档详解 #第一步:把sam文件转换成bam文件,我们得到map.bam文件 system"samtools view -bS m ...
- sam格式详细说明
原文链接 https://www.jianshu.com/p/386f520e5de1 The SAM Format Specification(sam格式说明) 1 The SAM Format S ...
- Picard报错“MAPQ should be 0 for unmapped read”的解决方法
picard对bwa生成的sam文件进行reorder时,报错如下: Getting Help Exception in thread "main" htsjdk.samtools ...
- kallisto:Near-optimal RNA-Seq quantification
Near-optimal RNA-Seq quantification https://pachterlab.github.io/kallisto 文章标题: Pseudoalignment fo ...
- GATK使用说明-GRCh38(Genome Reference Consortium)(二)
Reference Genome Components 1. GRCh38 is special because it has alternate contigs that represent pop ...
- 生物信息学工具--bowtie&bowtie2
Bowtie和Bowtie2使用 [怪毛匠子整理] Source URL: http://www.bbioo.com/lifesciences/40-112837-1.html Bowtie和Bowt ...
- Linux command line exercises for NGS data processing
by Umer Zeeshan Ijaz The purpose of this tutorial is to introduce students to the frequently used to ...
- HTseq-count
HTSeq:一个用于处理高通量数据(High-throughout sequencing)的python包.HTSeq包有很多功能类,熟悉python脚本的可以自行编写数据处理脚本.另外,HTSeq也 ...
- STAR manual
来源:STARmanual.pdf 来源:Calling variants in RNAseq PART0 准备工作 #STAR 安装前的依赖的工具 #Red Hat, CentOS, Fedora. ...
随机推荐
- POJ 1062:昂贵的聘礼
昂贵的聘礼 Time Limit: 1000MS Memory Limit: 10000K Total Submissions: 40715 Accepted: 11839 Descripti ...
- mnist lenet caffe 测试
# -*- coding: utf-8 -*-import sysimport numpy as npimport structfrom PIL import Imageprint "hel ...
- C++逐词读取txt
这一篇来写下std::ifstream读取txt的另一种方式,逐词读取,上一篇是按行读取,逐词读取的话每个单词都以空格或者换行等符号间隔开. 代码如下: #include "stdafx.h ...
- LeetCode 687. Longest Univalue Path 最长同值路径 (C++/Java)
题目: Given a binary tree, find the length of the longest path where each node in the path has the sam ...
- 吴裕雄--天生自然 JAVASCRIPT开发学习:Math(算数) 对象
<!DOCTYPE html> <html> <head> <meta charset="utf-8"> <title> ...
- 吴裕雄--天生自然 JAVASCRIPT开发学习: DOM
<!DOCTYPE html> <html> <head> <meta charset="utf-8"> <title> ...
- Nginx和php交互的两种方式
Unix socket 也叫IPC socket 也就是进程间通信套接字用于同一台主机上的不同进程间交换数据 TCP socket IP socket要利用主机的传输层(tcp),可以用于同一台主机 ...
- mysql字符集配置&mysql中文乱码
问题描述 这两天重置了下自己的电脑系统,一个ubuntu,另外一个当然就是windows. 不过在运行程序的时候发现,出现了很多的"????",也就是乱码字符.毫无疑问,这定然是m ...
- Python笔记_第四篇_高阶编程_再议装饰器和再议内置函数
1. 概述: 我们在前面用了很多的装饰器这个工具的方法.这个位置要系统的讲一下装饰器. 1.2 为什么需要装饰器. 装饰器本质是一个Python函数,它可以让其他函数在不需要任何代码变动的前提下增加额 ...
- Android studio中2种build.gradle文件介绍
根目录下的build.gradle通常不需要修改这个文件中的内容,除非需要添加一些全局的项目构建配置 buildscript { repositories { google() //声明代码托管仓库G ...