DEXSeq
1)Introduction
DEXSeq是一种在多个比较RNA-seq实验中,检验差异外显子使用情况的方法。 通过差异外显子使用(DEU),我们指的是由实验条件引起的外显子相对使用的变化。 外显子的相对使用定义为:
number of transcripts from the gene that contain this exon / number of all transcripts from the gene
大致思想:. For each exon (or part of an exon) and each sample, we count how many reads map to this exon and how many reads map to any of the other exons of the same gene. We consider the ratio of these two counts, and how it changes across conditions, to infer changes in the relative exon usage
2)安装
if("DEXSeq" %in% rownames(installed.packages()) == FALSE) {source("http://bioconductor.org/biocLite.R");biocLite("DEXSeq")}
suppressMessages(library(DEXSeq))
ls('package:DEXSeq')
pythonScriptsDir = system.file( "python_scripts", package="DEXSeq" )
list.files(pythonScriptsDir)
## [1] "dexseq_count.py" "dexseq_prepare_annotation.py" #查看是否含有这两个脚本
python dexseq_prepare_annotation.py Drosophila_melanogaster.BDGP5.72.gtf Dmel.BDGP5.25.62.DEXSeq.chr.gff #GTF转化为GFF with collapsed exon counting bins.
python dexseq_count.py Dmel.BDGP5.25.62.DEXSeq.chr.gff untreated1.sam untreated1fb.txt #count
3) 用自带实验数据集(数据预处理)
suppressMessages(library(pasilla))
inDir = system.file("extdata", package="pasilla")
countFiles = list.files(inDir, pattern="fb.txt$", full.names=TRUE) #countfile(如果不是自带数据集,可以由dexseq_count.py脚本生成)
basename(countFiles)
flattenedFile = list.files(inDir, pattern="gff$", full.names=TRUE)
basename(flattenedFile) #gff文件(如果不是自带数据集,可以由dexseq_prepare_annotation.py脚本生成)
########构造数据框sampleTable,包含sample名字,实验,文库类型等信息#######################
sampleTable = data.frame(
row.names = c( "treated1", "treated2", "treated3",
"untreated1", "untreated2", "untreated3", "untreated4" ),
condition = c("knockdown", "knockdown", "knockdown",
"control", "control", "control", "control" ),
libType = c( "single-end", "paired-end", "paired-end",
"single-end", "single-end", "paired-end", "paired-end" ) )
sampleTable ##############构建 DEXSeqDataSet object#############################
dxd = DEXSeqDataSetFromHTSeq(
countFiles,
sampleData=sampleTable,
design= ~ sample + exon + condition:exon,
flattenedfile=flattenedFile ) #四个参数
4)Standard analysis work-flow
########以下是简单的实验设计#####
genesForSubset = read.table(file.path(inDir, "geneIDsinsubset.txt"),stringsAsFactors=FALSE)[[1]] #基因子集ID
dxd = dxd[geneIDs( dxd ) %in% genesForSubset,] #取子集,减少运行量
head(colData(dxd))
head( counts(dxd), 5 )
split( seq_len(ncol(dxd)), colData(dxd)$exon )
sampleAnnotation( dxd )
############# dispersion estimates and the size factors#############
dxd = estimateSizeFactors( dxd ) ##Normalisation
dxd = estimateDispersions( dxd )
plotDispEsts( dxd ) #图1 #################Testing for differential exon usage############
dxd = testForDEU( dxd )
dxd = estimateExonFoldChanges( dxd, fitExpToVar="condition")
dxr1 = DEXSeqResults( dxd )
dxr1
mcols(dxr1)$description
table ( dxr1$padj < 0.1 )
table ( tapply( dxr1$padj < 0.1, dxr1$groupID, any ) )
plotMA( dxr1, cex=0.8 ) #图2

To see how the power to detect differential exon usage depends on the number of reads that map to an exon, a so-called MA plot is useful, which plots the logarithm of fold change versus average normalized count per exon and marks by red colour the exons which are considered significant; here, the exons with an adjusted p values of less than 0.1

############以下是更复杂的实验设计##################
formulaFullModel = ~ sample + exon + libType:exon + condition:exon
formulaReducedModel = ~ sample + exon + libType:exon
dxd = estimateDispersions( dxd, formula = formulaFullModel )
dxd = testForDEU( dxd,
reducedModel = formulaReducedModel,
fullModel = formulaFullModel )
dxr2 = DEXSeqResults( dxd )
table( dxr2$padj < 0.1 )
table( before = dxr1$padj < 0.1, now = dxr2$padj < 0.1 )##和简单的实验设计比较
5)Visualization
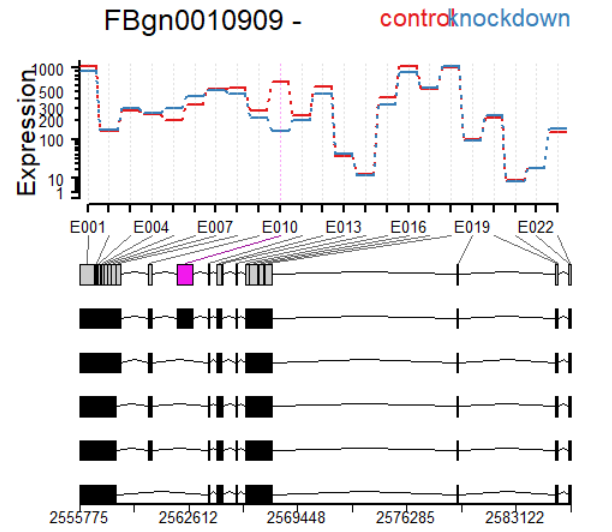
plotDEXSeq( dxr2, "FBgn0010909", legend=TRUE, cex.axis=1.2, cex=1.3,
lwd=2 )
plotDEXSeq( dxr2, "FBgn0010909", displayTranscripts=TRUE, legend=TRUE,
cex.axis=1.2, cex=1.3, lwd=2 )
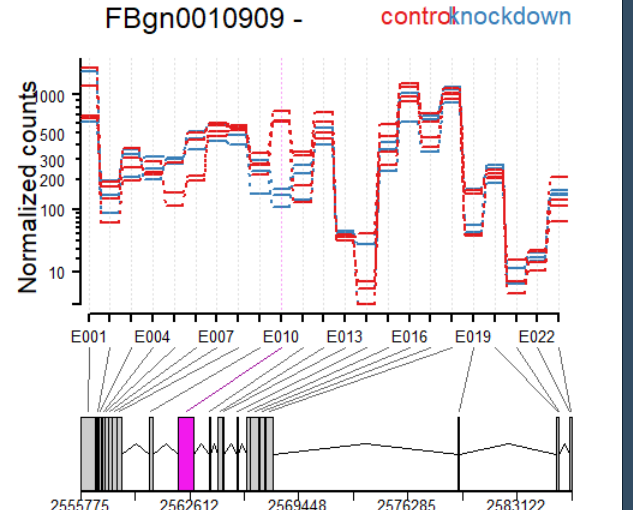
plotDEXSeq( dxr2, "FBgn0010909", expression=FALSE, norCounts=TRUE,
legend=TRUE, cex.axis=1.2, cex=1.3, lwd=2 )
plotDEXSeq( dxr2, "FBgn0010909", expression=FALSE, splicing=TRUE,
legend=TRUE, cex.axis=1.2, cex=1.3, lwd=2 )
DEXSeqHTML( dxr2, FDR=0.1, color=c("#FF000080", "#0000FF80") )


DEXSeq的更多相关文章
- 【转录组入门】6:reads计数
作业要求: 实现这个功能的软件也很多,还是烦请大家先自己搜索几个教程,入门请统一用htseq-count,对每个样本都会输出一个表达量文件. 需要用脚本合并所有的样本为表达矩阵.参考:生信编程直播第四 ...
- Bulk RNA-Seq转录组学习
与之对应的是single cell RNA-Seq,后面也会有类似文章. 参考:https://github.com/xuzhougeng/Learn-Bioinformatics/ 作业:RNA-s ...
- Bioconductor应用领域之基因芯片
引用自https://mp.weixin.qq.com/s?__biz=MzU4NjU4ODQ2MQ==&mid=2247484662&idx=1&sn=194668553f9 ...
随机推荐
- 关于buffer,cache,wb,wt,clean,inv,flush,以及其他
1. 有时候需要区分buffer和cache:buffer解决CPU写的问题,比如将多次写操作buffer起来一次性更新:cache解决CPU读的问题,将数据cache起来在下次读的时候快速取用. 2 ...
- BASIC-9_蓝桥杯_特殊回文数
示例代码: #include <stdio.h> int main(void){ int n = 0 ; scanf("%d",&n); int i = 0 ; ...
- centos7开机界面出现多个选项
第一个选项正常启动,第二个选项急救模式启动(系统出项问题不能正常启动时使用并修复系统) 在CentOS更新后,并不会自动删除旧内核.所以在启动选项中会有多个内核选项,可以手动使用以下命令删除多余的内核 ...
- pyplot 绘图与可视化
1. 基本使用 #!/usr/bin/env python # coding=utf-8 import matplotlib.pyplot as plt from numpy.random impor ...
- Linux下几种反弹Shell方法的总结与理解
之前在网上看到很多师傅们总结的linux反弹shell的一些方法,为了更熟练的去运用这些技术,于是自己花精力查了很多资料去理解这些命令的含义,将研究的成果记录在这里,所谓的反弹shell,指的是我们在 ...
- Selenium如何定位动态id的元素?
怎么定位这类型的元素呢? 根据其他属性定位如果有其他固定属性,最先考虑的当然是根据元素的其他属性来定位,定位方式那么多,何必在这一棵树上吊死.. 根据相对关系定位根据其附近的父节点.子节点.兄弟节点定 ...
- Linux 各类设置、配置、使用技巧参考,Linux使用集锦
========== 参考格式 (新增记录时,复制粘贴在下)============= [日期]: <标题> 参考链接ref1: 参考链接ref2: 正文: ========== 参考格式 ...
- (15/24) 为webpack增加babel支持
Babel是什么? Babel是一个编译JavaScript的平台,它的强大之处表现在可以通过编译达到以下目的: 使用下一代的javaScript代码(ES6,ES7-.),即使这些标准目前并未被当前 ...
- THREE.JS 场景世界坐标和平面二维坐标互转
<!DOCTYPE html><html lang="en"><head> <meta charset="UTF-8&qu ...
- java.lang.ClassCastException: java.util.Arrays$ArrayList cannot be cast to java.util.ArrayList
String[] 转换成 ArrayList 报的错. String[] str = {"A","B"}; ArrayList<String> li ...