Data-independent acquisition mass spectrometry in metaproteomics of gut microbiota - implementation and computational analysis DIA技术在肠道宏蛋白质组研究中的方法实现和数据分析 (解读人:闫克强)
文献名:Data-independent acquisition mass spectrometry in metaproteomics
of gut microbiota - implementation and computational analysis(DIA技术在肠道宏蛋白质组研究中的方法实现和数据分析)
doi: 10.1021/acs.jproteome.9b00606
期刊名:Journal of Proteome Research
作者:Juhani Akakko, Sami Pietila
通讯作者: Laura L. Elo
单位:
- 奥博学术大学
一、 概述:
在宏蛋白质组的研究中,目前主要的研究方法是基于DDA(数据依赖型采集模式)的方法进行蛋白的鉴定和非标定量。但对于复杂的环境样本来讲,这种定量方法存在较低的重复性。为了解决这个问题,在本研究中,采用了DIA(非数据依赖型采集模式)对宏蛋白质组的样本进行分析。同时,开发了用于分析宏蛋白质组DIA数据的软件包diatools。通过构建的模拟微生物混合样本以及人粪便样本验证了该方法的适用性。
二、 研究背景:
宏蛋白质组学研究可以从功能的角度来揭示微生物群落在其生态系统中的功能。目前,主流的宏蛋白质组研究方法是基于DDA的方法。但该方法的样本重复性会随着样本复杂度的上升而降低,定量结果具有高的偏好性。因此并不适合于宏蛋白质样本的研究。DIA的方法可以有效解决这些问题,但目前为止,DIA方法还未用于肠道微生物的宏蛋白质组研究,同时也缺乏相应的分析工具对这样的数据进行解析。
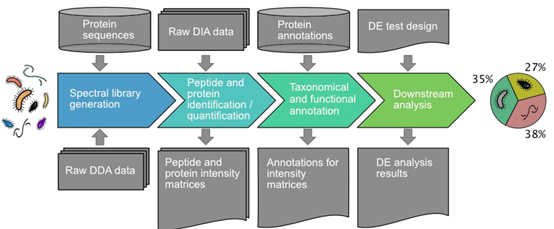
本研究建立了一套利用DIA方法对环境微生物宏蛋白质组样本进行分析的流程,同时开发了用于解析该类数据的工具包diatools。
Diatools的数据处理主要分两个步骤:1)利用DDA数据产生谱图库2)利用构建好的谱图库对DIA数据进行鉴定和定量。该软件包可以在github上进行下载和安装(https://github.com/elolab/diatools)。
三、 实验设计:
本研究主要分两个阶段。一是宏蛋白质组DIA软件包的构建,二是对该方法的评估。
Diatools软件包
该软件包将质谱上机的raw data作为输出,输出为肽段对应强度的矩阵。简单来讲,该软件利用DDA数据构建谱图库,然后利用该结果对DIA数据进行鉴定和定量。其中包含了很多标准的蛋白分析工具和流程,用于支持对复杂样本的数据分析。包括OpenMS,Trans-Proteomic Pipeline (TPP),msproteomicstools等。
微生物群落样本
为了对该流程进行验证,本研究选取了两类微生物群落样本,一类是模拟环境样本的混合样本。包含了12个菌株,每个菌株取等量细胞的量进行混合。另一类是6个人粪便样本。
蛋白数据分析
在DDA层面,采用X!tandem和Comet对DDA数据进行搜库鉴定,数据库为人肠道微生物数据库,共包含9.9百万条蛋白序列。肽段水平FDR设为1%。在DIA层面,利用DDA构建的谱图库进行DIA数据的解析,实现对肽段的定量。物种和功能注释皆来源于数据库信息。
四、研究成果:
Fig1A和B分别展示了3个技术重复在肽段鉴定和定量层面的情况。可以看出,3组技术重复无论是在鉴定还是在定量层面都非常接近,这说明利用DIA的方法的确会得到非常好的样本重复性。Fig1C展示了肽段在属层面的鉴定比例,约43%的肽段可以归并到属层级,大部分肽段在属层面的归并还是模糊的。

Fig2A展示了6个人粪便样本的在肽段鉴定水平上的相似性,Fig2B比较了DIA和DDA在肽段鉴定水平上的差异。可以看出,DIA的肽段鉴定相似度远高于DDA水平。Fig2C展示了粪便样本的物种归并结果。同样的,大部分肽段在属层级的归并较模糊。

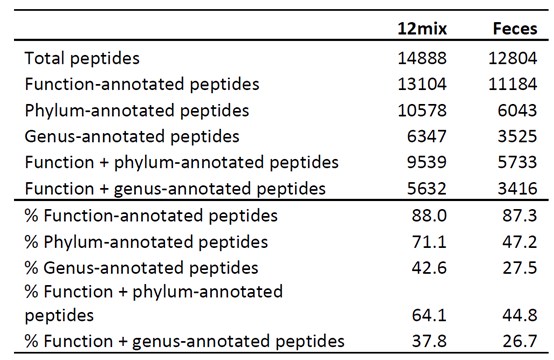
表1展示了两类样本的鉴定情况,包括物种鉴定和功能鉴定结果。在肽段功能注释结果中,KOG的注释率在两个样本中均比较高,这说明由肽段去推断微生物的功能可能是比较好的方法,这将避免在蛋白归并中可能出现的一些错误。
五、文章评论:
本文结合了DIA的方法来对宏蛋白质组样本进行分析,并建立了一套数据分析流程,并取得了比较好的结果。该为接下来的宏蛋白质组研究提供了新的思路。但本研究仅从样本定量重复性以及肽段鉴定重复性方面进行了评估,评估方法过于简单,无法真正凸显DIA数据的优势。此外,该DIA数据分析流程与常规DIA分析方法并无显著差异,没有针对宏蛋白质组数据的特征进行优化。
阅读人:闫克强
Data-independent acquisition mass spectrometry in metaproteomics of gut microbiota - implementation and computational analysis DIA技术在肠道宏蛋白质组研究中的方法实现和数据分析 (解读人:闫克强)的更多相关文章
- Systematic comparison of strategies for the enrichment of lysosomes by data independent acquisition 通过DIA技术系统比较各溶酶体富集策略 (解读人:王欣然)
文献名:Systematic comparison of strategies for the enrichment of lysosomes by data independent acquisit ...
- Fast and accurate bacterial species identification in urine specimens using LC-MS/MS mass spectrometry and machine learning (解读人:闫克强)
文献名:Fast and accurate bacterial species identification in urine specimens using LC-MS/MS mass spectr ...
- MCP|LQD|Data-independent acquisition improves quantitative cross-linking mass spectrometry (DIA方法可提升交联质谱定量分析)
文献名:Data-independent acquisition improves quantitative cross-linking mass spectrometry (DIA方法可提升定量交联 ...
- Journal of Proteome Research | Single-Shot Capillary Zone Electrophoresis−Tandem Mass Spectrometry Produces over 4400 Phosphopeptide Identifications from a 220 ng Sample (分享人:赵伟宁)
Title: Single-Shot Capillary Zone Electrophoresis−Tandem Mass Spectrometry Produces over 4400 Phosph ...
- MCP|DYM|Quantitative mass spectrometry to interrogate proteomic heterogeneity in metastatic lung adenocarcinoma and validate a novel somatic mutation CDK12-G879V (利用定量质谱探究转移性肺腺瘤的蛋白质组异质性及验证新体细胞突变)
文献名:Quantitative mass spectrometry to interrogate proteomic heterogeneity in metastatic lung adenoca ...
- Development of a High Coverage Pseudotargeted Lipidomics Method Based on Ultra-High Performance Liquid Chromatography−Mass Spectrometry(基于超高效液相色谱-质谱法的高覆盖拟靶向脂质组学方法的开发)
文献名:Development of a High Coverage Pseudotargeted Lipidomics Method Based on Ultra-High Performance ...
- 关于Jquery中ajax方法data参数用法的总结
data 发送到服务器的数据.将自动转换为请求字符串格式.GET 请求中将附加在 URL 后.查看 processData 选项说明以禁止此自动转换.必须为 Key/Value 格式.如果为数组,jQ ...
- Comparing Data-Independent Acquisition and Parallel Reaction Monitoring in Their Abilities To Differentiate High-Density Lipoprotein Subclasses 比较DIA和PRM区分高密度脂蛋白亚类的能力 (解读人:陈凌云)
文献名:Comparing Data-Independent Acquisition and Parallel Reaction Monitoring in Their Abilities To Di ...
- Journal of Proteome Research | Utilization of the Proteome Data Deposited in SRMAtlas for Validating the Existence of the Human Missing Proteins in GPM (解读人:梁嘉琪)
文献名:Utilization of the Proteome Data Deposited in SRMAtlas for Validating the Existence of the Human ...
随机推荐
- js 原生轮播图插件
<!doctype html> <html lang="en"> <head> <meta charset="UTF-8&quo ...
- Jenkins之邮件通知
Jenkins默认有一个邮件通知功能,但功能比较单一,能自定义的东西很少,一般使用Extended E-mail Notification插件来发送邮件,EXT Email插件功能比较多,但设置也比较 ...
- SWUST OJ Delete Numbers(0700)
Delete Numbers(0700) Time limit(ms): 1000 Memory limit(kb): 65535 Submission: 1731 Accepted: 373 D ...
- Parentheses Balance (括号平衡)---栈
题目链接:https://vjudge.net/contest/171027#problem/E Yes的输出条件: 1. 空字符串 2.形如()[]; 3.形如([])或者[()] 分析: 1.设置 ...
- js作用域其二:预解析
文章目錄 解析机制 JavaScript是一门解释型的语言 , 想要运行js代码需要两个阶段 编译阶段: 编译阶段就是我们常说的JavaScript预解析(预处理)阶段,在这个阶段JavaScript ...
- charles添加https支持
- iOS 使用系统的UITabBarController 修改展示的图片大小
1. 设置TabBarItem图片的大小 1 - (void)configurationAppTabBarAndNavigationBar { // 选中的item普通状态图片的大小 UIImage ...
- Web渗透基础小总结
Web渗透框架概述 主要组成: 1. web语言代码(脚本) 2. web程序 3. 数据库程序 Web语言常见几大类 1. HTML:超文本标记语言,标准通用编辑语言下的一个应用 2. PHP:超文 ...
- 密码学习(一)——Base64
简介 Base64是一种非常常用的数据编码方式,标准Base64可以把所有的数据用"A~Z,a~z,0~9,+,/,="共65个字符(‘=’号仅是一个占位符,作为后缀)表示,当然在 ...
- IDEA中Git的使用详解
原文链接:https://www.cnblogs.com/javabg/p/8567790.html 工作中多人使用版本控制软件协作开发,常见的应用场景归纳如下: 假设小组中有两个人,组长小张,组员小 ...