RNA-seq连特异性
RNA-seq连特异性
Oct 15, 2015
The strandness of RNA-seq analysis
前段时间一直在研究关于illumina TrueSeq stranded RNA-seq中的strand如何判断的问题。之后我又查了很多资料,终于弄懂了。现在写下来,如果我有错误,欢迎继续指正。 以下文字和图片的引用链接都已经给出,如果图片在邮件中无法显示,可以打开链接的。
先说结论:对于Illumina TrueSeq stranded RNA protocol,主要采用的是dUTP method。RNA的strand是与read1(先测的read)相反,与read2(后测的read)的strand相同的。换句话说,如果read1比对到基因组正链上,则对应的RNA是在基因组的负链上。如果read2比对到基因组正链上,则对应的RNA应该是比对到正链上。
具体解释如下:
1.关于strand:
1)DNA是由两条互补配对的链组成的。按照预定俗称,把其中一条链称为正链(plus strand or forward strand),另一条则是负链(minus strand or reverse strand),这个定义完全是人为的。我们下载的hg18、hg19、hg38的fasta格式只给出了正链的序列。另一条链可以根据互补配对得出。
2)我们读取的链的时候,经常是从5’到3’的,无论DNA还是RNA。测序出来的两个read,也都是从5’到3’的。视觉上,我们读取plus strand的时候,from left to right。读取minus strand的时候,from right to left。
3)RNA会根据一条DNA模板合成,我们在IGV或者UCSC上会看到RNA的链的信息,如果一条RNA在正链上(如下图),其实该条RNA是以负链作为模板形成的,但是该条RNA与正链的序列一直。我们把和合成RNA时的DNA模板的链称为template strand或者antisense strand。把与RNA序列相同的DNA的链称为coding strand或者sense strand。sense/antisense, coding/template strand可以在基因组的plus strand也可以在基因组的minus strand。
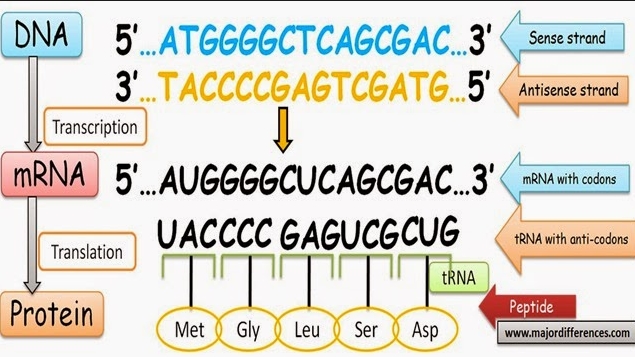
http://www.majordifferences.com/2015/01/difference-between-sense-and-antisense.html
2、illumina测序之sample preparation:
1)illumina测序的sample preparation的步骤,一般是讲DNA或者是cDNA打断,然后repair ends,add A at 3’end (为了和Y-shaped adaptor结合),然后加上adaptor。
2)adaptor也是由两条链组成的(universal adaptor和indexed adaptor)。unversal adaptor的3’端有一个T,为了可以和DNA fragment上的A互补。universal adaptor的3’端和indexed adaptor的5’端有12bp的碱基互补配对,使得他们两个可以呈现Y字形。indexed adaptor的中间有6个bp的indexed sequence,可以用来在同一个land中有多个样本时做区分。
3)若是非连特异性的,对于最后合成的sample,中间的DNA fragment既可以来自正链,也可以来自负链。
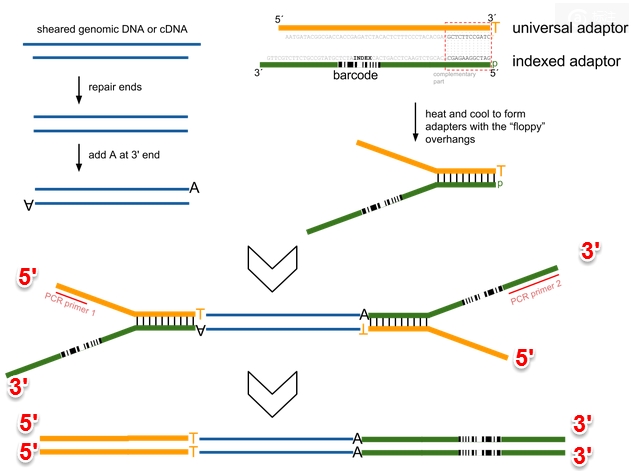
http://onetipperday.blogspot.com/2013/06/illumina-hiseq2000-adaptor.html
3、dUTP method的sample preparation:
dUTP metod主要是在反转完first-strand cDNA之后,在合成second-strand cDNA的时候,用dUTP代替了dTTP,之后加完adaptor,用UDGase处理,将second-strand cDNA消化掉。这样最后合成的sample,中间的DNA fragment一定是first-strand cDNA。这也是为什么对于dUTP数据处理,tophat的参数应该为“–library-type fr-firststrand”。注意,这里的first-strand cDNA与RNA strand(或者基因组上的coding strand)相反。
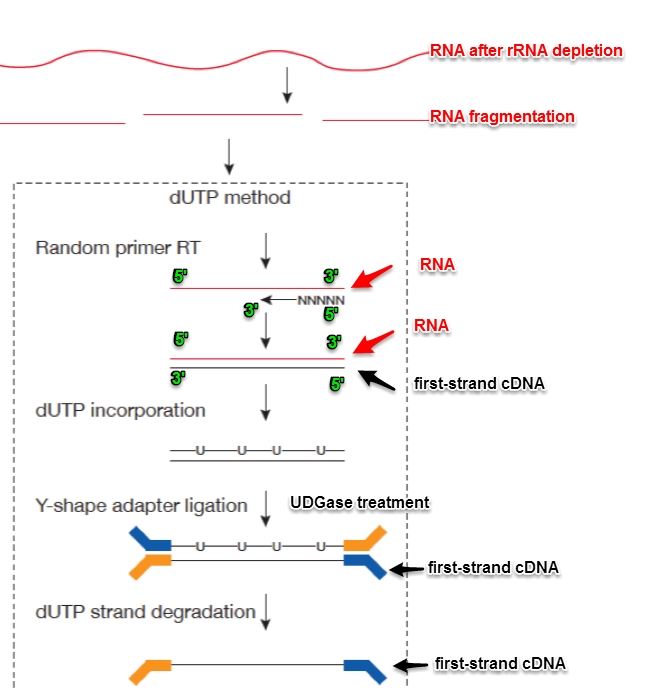
http://onetipperday.blogspot.com/2013/06/illumina-hiseq2000-adaptor.html
4、illumina测序之cluster generation和sequencing
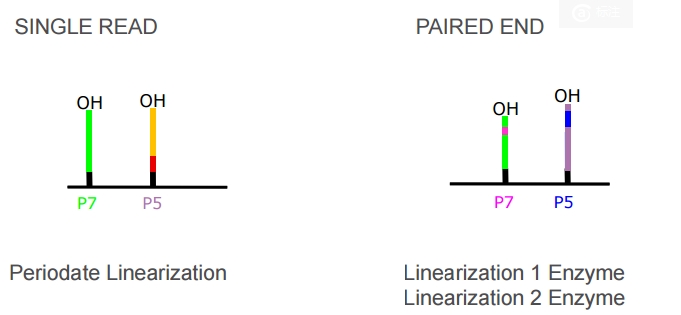
1)sample preparation之后,样品就上机测序了。在flow cell的表面,固定着很多个DNA序列,他们有两种P5和P7,均是5’在flow cell的表面,3’露在上面。
2)P5的序列与sample 5’端的unversial adapter 开始的44(数字可以不同)个bases相同,P7的是与sample的3’端的indexed adapter的24(数字可以不同)个bases互补配对。P5和P7同时可以充当与sample DNA序列杂交的探针还有PCR扩增的引物。
3)sample DNA上机后,首先与P7杂交,然后以P7为引物合成。如下图,之后洗掉原来的sample DNA,然后P7上的DNA的3’端正好与P5杂交,建起bright,然后再以P5为引物合成。就这样不断amplification。
4)之后测序的时候,会先将P5进行Periodate linearization(具体不是很懂,应该就是把P5上的DNA弄走)。然后用引物来测定P7中间DNA fragment的序列,就是read1中的序列(注意,这里的引物与P5和P7不同,可以保证直接测出中间感兴趣的序列。但是若中间DNA fragement太短,就会测到adaptor序列,这就是为什么fastq文件要做adaptor trimming)。如果是Pair-end序列。P7上的序列测完之后,再合成P5的DNA,然后再用另一种酶把P7弄走,去测P5的序列。这样就保证测量了pair-end。从这个过程可以看到read1和read2一定是分布会比对到基因组不同的链上。
5)对于strand-specifc,由于sample DNA中间感兴趣的DNA fragement只有first-strand。这个fragment先于P7杂交,然后合成second strand cDNA。然后测序的时候,read1是与P7上的second strand cDNA互补配对的。(first-strand cDNA <- rc -> P7上的second strand cDNA <-rc->read1的序列)。故read1与first-strand cDNA的链相同,与RNA的strand链相反。反之,read2与RNA strand的链相同。
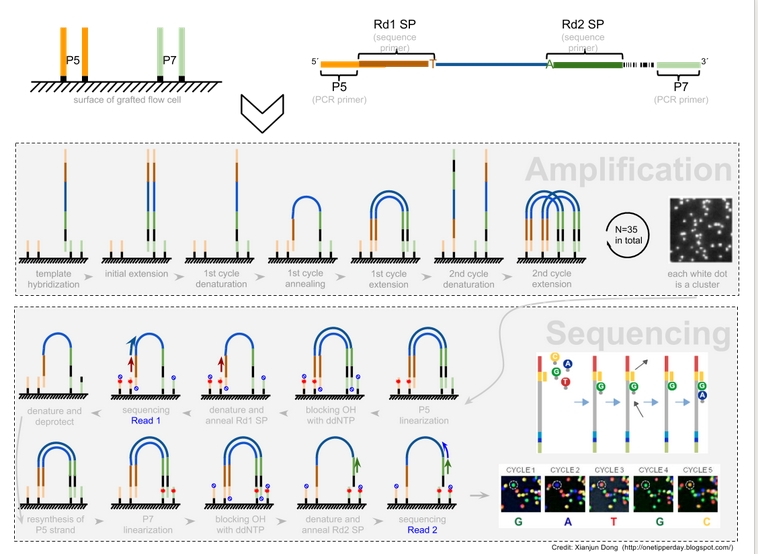
http://onetipperday.blogspot.com/2013/12/illumina-hiseq2000-adaptor-and.html
http://skatebase.org/sites/skatebase.org/files/workshops/W1_IntroIlluminaNGS.pdf
5、tophat中output的bam文件中有一个tag XS是错的
tophat输出的bam文件的attribute tag中有一个XS是指示RNA所在的strand的。但是很多人都发现了错误。而且Encode还写了一个perl脚本来修改XS tag。也有人说XS主要是根据splite site的序列判断的。XS: “+” for GT-AG and “-“ for CT-AC。另外,因为cufflinks是需要XS的这个tag的,所以这个问题还需要再研究一下。
6、可以通过sam文件的第2列的flag来判断read的strand
sam flag可以指示该条read是read1(first in pair)还是read2(second in pair),或者比对到正链上(mate reverse strand)还是负链上(read reverse strand)。 我其实也不懂得flag这个16进制,这有个网址给了一个简单的软件,可以帮助计算。然后用samtools view -f或者-F就可以根据flag来筛选了。
7、参考资料:
http://onetipperday.blogspot.com/2013/06/illumina-hiseq2000-adaptor.html
http://onetipperday.blogspot.com/2012/07/how-to-tell-which-library-type-to-use.html
http://onetipperday.blogspot.com/2013/12/illumina-hiseq2000-adaptor-and.html
http://www.personal.psu.edu/iua1/courses/illumina-sequencing.html
http://skatebase.org/sites/skatebase.org/files/workshops/W1_IntroIlluminaNGS.pdf
RNA-seq连特异性的更多相关文章
- RNA seq 两种计算基因表达量方法
两种RNA seq的基因表达量计算方法: 1. RPKM:http://www.plob.org/2011/10/24/294.html 2. RSEM:这个是TCGAdata中使用的.RSEM据说比 ...
- RNA -seq
RNA -seq RNA-seq目的.用处::可以帮助我们了解,各种比较条件下,所有基因的表达情况的差异. 比如:正常组织和肿瘤组织的之间的差异:检测药物治疗前后,基因表达的差异:检测发育过程中,不同 ...
- RNA测序相对基因表达芯片有什么优势?
RNA测序相对基因表达芯片有什么优势? RNA-Seq和基因表达芯片相比,哪种方法更有优势?关键看适用不适用.那么RNA-Seq适用哪些研究方向?是否您的研究?来跟随本文了解一下RNA测序相对基因表达 ...
- featureCounts 软件说明
featuresCounts 软件用于定量,不仅可以支持gene的定量,也支持exon, gene bodies, genomic bins, chromsomal locations的定量: 官网 ...
- Advances in Single Cell Genomics to Study Brain Cell Types | 会议概览
单细胞在脑科学方面的应用 Session 1: Deciphering the Cellular Landscape of the Brain Using Single Cell Transcript ...
- xgene:WGS,突变与癌,RNA-seq,WES
人类全基因组测序06 SNP(single nucleotide polymorphism):有了10倍以上的覆盖深度以后,来确认SNP信息,就相当可靠了. 一个普通黄种人的基因组,与hg19这个参 ...
- 链终止法|边合成边测序|Bowtie|TopHat|Cufflinks|RPKM|FASTX-Toolkit|fastaQC|基因芯片|桥式扩增|
生物信息学 Sanger采用链终止法进行测序 带有荧光基团的ddXTP+其他四种普通的脱氧核苷酸放入同一个培养皿中,例如带有荧光基团的ddATP+普通的脱氧核苷酸A.T.C.G放入同一个培养皿,以此类 ...
- xgene:之ROC曲线、ctDNA、small-RNA seq、甲基化seq、单细胞DNA, mRNA
灵敏度高 == 假阴性率低,即漏检率低,即有病人却没有发现出来的概率低. 用于判断:有一部分人患有一种疾病,某种检验方法可以在人群中检出多少个病人来. 特异性高 == 假阳性率低,即错把健康判定为病人 ...
- RNA提取和建库流程对mRNA-Seq的影响
RNA提取和建库流程对mRNA-Seq的影响 已有 10460 次阅读 2014-8-14 14:21 |个人分类:转录组测序|系统分类:科研笔记|关键词:转录组测序,RNA-Seq,,链特异性RNA ...
- RNA Spike-in Control(转)
Spike-in Control:添加/加入(某种物质)的对照(组)在某些情况下,待检验样本中不含待测物质或者含有但是浓度很低,为了证明自己建立的方法能对样本中待测物质进行有效的检测,可在待检样本中加 ...
随机推荐
- Oracle 存储过程 延迟执行 DBMS_LOCK.SLEEP(60);
--测试代码: declare -- Local variables here i integer; begin -- Test statements here dbms_output.put_l ...
- flask框架预备知识
1.web预备知识 2.flask介绍 3.web框架的本质及分类 4.flask安装与基本设置 1.web预备知识 HTTP协议:https://www.cnblogs.com/wyb666/p/9 ...
- windows10系统telnet登陆
参考网站:https://jingyan.baidu.com/article/acf728fd498e9ff8e510a322.html windows10系统以及没有telnet服务器端了. 需要单 ...
- sbt的安装测试
1.下载 wget https://github.com/sbt/sbt/releases/download/v0.13.15/sbt-0.13.15.tgz 2.安装 tar -zxvf sbt-0 ...
- HTML5 Canvas 小例子 旋转的图片
<一>CSS部分 @charset "utf-8"; *{ padding:; margin:; outline: none; } #canvas{ position: ...
- C# 生成word文档(NPOI)
using NPOI.XWPF.UserModel XWPFDocument doc = new XWPFDocument(); //创建新的word文档 XWPFParagraph p1 = doc ...
- 1.java生成随机不重复10位字符串
package org.changneng.util; import java.util.Random; public class A { public static void main(String ...
- mybatis 常用jdbcType数据类型
来自 : http://blog.csdn.net/paincupid/article/details/50922981
- Eclipse 更改Maven项目名
1. 在Eclipse 中修改项目名 没错这种方法跟你预料的一样简单,当项目已经导入到 Eclipse 之后,只需要做两个事情 1.1 改项目文件夹名称 选中项目,按 F12 ,改名. 多数人改项目名 ...
- 6 unit3-文件操作&函数 review
文件处理相关 1.编码问题 (1)请说明python2 与python3中的默认编码是什么? py2默认ASCII码,py3默认的utf8 (2)为什么会出现中文乱码?你能列举出现乱码的情况有哪几种? ...