GSVA的使用
- GSVA的简介
Gene Set Variation Analysis,被称为基因集变异分析,是一种非参数的无监督分析方法,主要用来评估芯片核转录组的基因集富集结果。主要是通过将基因在不同样品间的表达量矩阵转化成基因集在样品间的表达量矩阵,从而来评估不同的代谢通路在不同样品间是否富集。其实就是研究这些感兴趣的基因集在不同样品间的差异,或者寻找比较重要的基因集,作为一种分析方法,主要是是为了从生物信息学的角度去解释导致表型差异的原因。它的主要输入文件为表达量的矩阵和基因集的文件,通过gsva的方法就可以得出结果;既可以处理芯片的结果,也可以处理转录组的结果。 - GSVA的安装
source("http://bioconductor.org/biocLite.R")
biocLite('GSVAdata')
biocLite('GSVA')
biocLite("limma")
biocLite("genefilter")
- GSVA的运行注意事项
- 如果是芯片的数据是需要对数据进行过滤的,可以参考下面的测试数据代码,或者看官方的文档
- GSVA本身提供了三个算法,一般使用默认的算法就可以了
- 对于RNA-seq的数据,如果是read count可以选择Possion分布,如果是均一化后的表达量值,可以选择默认参数高斯分布就可以了
- 读入的数据格式可以参考最后的表达谱数据格式,需要自己制作分组信息
- 模拟数据的使用
#构造一个 30个样本,2万个基因的表达矩阵, 加上 100 个假定的基因集
library(GSVA)
p <- 20000 ## number of genes
n <- 30 ## number of samples
nGS <- 100 ## number of gene sets
min.sz <- 10 ## minimum gene set size
max.sz <- 100 ## maximum gene set size
X <- matrix(rnorm(p*n), nrow=p, dimnames=list(1:p, 1:n))
dim(X)
gs <- as.list(sample(min.sz:max.sz, size=nGS, replace=TRUE)) ## sample gene set sizes
gs <- lapply(gs, function(n, p) sample(1:p, size=n, replace=FALSE), p) ## sample gene sets
es.max <- gsva(X, gs, mx.diff=FALSE, verbose=FALSE, parallel.sz=1)
es.dif <- gsva(X, gs, mx.diff=TRUE, verbose=FALSE, parallel.sz=1)
pheatmap::pheatmap(es.max)
pheatmap::pheatmap(es.dif)
- GSVA的使用
需要如下的包
library(GSEABase)
library(GSVAdata)
data(c2BroadSets)
c2BroadSets
library(Biobase)
library(genefilter)
library(limma)
library(RColorBrewer)
library(GSVA)
#官方文档有数据的预处理过程
cacheDir <- system.file("extdata", package="GSVA")
cachePrefix <- "cache4vignette_"
file.remove(paste(cacheDir, list.files(cacheDir, pattern=cachePrefix), sep="/"))
data(leukemia)
leukemia_eset
head(pData(leukemia_eset))
table(leukemia_eset$subtype)
- 过滤
data(leukemia)
leukemia_eset
filtered_eset <- nsFilter(leukemia_eset, require.entrez=TRUE, remove.dupEntrez=TRUE,var.func=IQR, var.filter=TRUE, var.cutoff=0.5, filterByQuantile=TRUE,feature.exclude="^AFFX")
leukemia_filtered_eset <- filtered_eset$eset
- GSVA的计算差异基因集
cache(leukemia_es <- gsva(leukemia_filtered_eset, c2BroadSets,min.sz=10, max.sz=500, verbose=TRUE),dir=cacheDir, prefix=cachePrefix)
adjPvalueCutoff <- 0.001
logFCcutoff <- log2(2)
design <- model.matrix(~ factor(leukemia_es$subtype))
colnames(design) <- c("ALL", "MLLvsALL")
fit <- lmFit(leukemia_es, design)
fit <- eBayes(fit)
allGeneSets <- topTable(fit, coef="MLLvsALL", number=Inf)
DEgeneSets <- topTable(fit, coef="MLLvsALL", number=Inf,
p.value=adjPvalueCutoff, adjust="BH")
res <- decideTests(fit, p.value=adjPvalueCutoff)
summary(res)
- limma计算差异表达基因
logFCcutoff <- log2(2)
design <- model.matrix(~ factor(leukemia_eset$subtype))
colnames(design) <- c("ALL", "MLLvsALL")
fit <- lmFit(leukemia_filtered_eset, design)
fit <- eBayes(fit)
allGenes <- topTable(fit, coef="MLLvsALL", number=Inf)
DEgenes <- topTable(fit, coef="MLLvsALL", number=Inf,
p.value=adjPvalueCutoff, adjust="BH", lfc=logFCcutoff)
res <- decideTests(fit, p.value=adjPvalueCutoff, lfc=logFCcutoff)
#summary(res)
- RNAseq的数据
如果是RNA-seq的原始整数的read count 在使用gsva时需要设置kcdf="Possion",如果是取过log的RPKM,TPM等结果可以使用默认的值,所以如果我们在使用fpkm进行分析的时候使用默认参数局可以了
data(commonPickrellHuang)
stopifnot(identical(featureNames(huangArrayRMAnoBatchCommon_eset),featureNames(pickrellCountsArgonneCQNcommon_eset)))
stopifnot(identical(sampleNames(huangArrayRMAnoBatchCommon_eset),sampleNames(pickrellCountsArgonneCQNcommon_eset)))
canonicalC2BroadSets <- c2BroadSets[c(grep("^KEGG", names(c2BroadSets)),grep("^REACTOME", names(c2BroadSets)),grep("^BIOCARTA", names(c2BroadSets)))]
data(genderGenesEntrez)
MSY <- GeneSet(msYgenesEntrez, geneIdType=EntrezIdentifier(),collectionType=BroadCollection(category="c2"), setName="MSY")
XiE <- GeneSet(XiEgenesEntrez, geneIdType=EntrezIdentifier(),collectionType=BroadCollection(category="c2"), setName="XiE")
canonicalC2BroadSets <- GeneSetCollection(c(canonicalC2BroadSets, MSY, XiE))
#使用GSVA方法进行计算
esmicro <- gsva(huangArrayRMAnoBatchCommon_eset, canonicalC2BroadSets, min.sz=5, max.sz=500,mx.diff=TRUE, verbose=FALSE, parallel.sz=1)
esrnaseq <- gsva(pickrellCountsArgonneCQNcommon_eset, canonicalC2BroadSets, min.sz=5, max.sz=500,kcdf="Poisson", mx.diff=TRUE, verbose=FALSE, parallel.sz=1)
- 其实文档中还比较了GSVA和其他不同方法,这里就略过了
实际的表达谱项目数据测试
表达量矩阵格式
gene AdA3_0h-1 AdA3_0h-2 AdA3_0h-3 AdA3_0h-4 AdA3_0h-5 AdA3_0h-6 AdA3_16h-3 AdA3_16h-4 AdA3_16h-5 AdA3_16h-6 AdGFP_0h-1 AdGFP_0h-2 AdGFP_0h-3 AdGFP_0h-4 AdGFP_0h-5 AdGFP_0h-6 AdGFP_16h-1 AdGFP_16h-2 AdGFP_16h-3 AdGFP_16h-4 AdGFP_16h-5 AdGFP_16h-6
Gnai3 73.842026 52.385326 73.034203 70.679092 67.094292 74.611313 74.853683 72.340401 73.230095 77.39888 70.019714 69.995277 72.172127 72.398514 74.176201 72.700516 60.29203 60.199333 58.043304 58.425671 61.812901 60.219734
Pbsn 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0
Cdc45 3.653137 5.67695 3.560387 3.287101 4.034892 2.92406 11.427282 14.665288 11.368333 11.96515 4.289696 3.869604 4.055799 3.617629 4.800964 4.790279 20.065876 25.753405 19.732252 26.296944 21.906717 20.378906
Scml2 0.15765 0.022811 0.301977 0.056921 0.022823 0.104651 1.542295 0.370989 0.226674 0.426919 0.132548 0.028549 0.066578 0 0.069393 0.10476 0.981483 1.607109 0.84421 0.912878 1.281105 2.309552
基因集格式
G1/S-Specific Transcription NA Rrm2 Ccne1 E2f1 Fbxo5 Cdc6 Dhfr Cdc45 Pola1 Cdt1 Cdk1 Tyms
E2F mediated regulation of DNA replication NA Rrm2 Ccne1 E2f1 Fbxo5 Cdc6 Pola2 Dhfr Cdc45 Pola1 Cdt1 Cdk1 Ccnb1 Tyms
Formation of Fibrin Clot (Clotting Cascade) NA Fga F11 Fgb Proc Serping1 Thbd Tfpi F7 Serpine2 Fgg Serpind1 Serpina5 F8 Pf4
Resolution of D-loop Structures through Holliday Junction Intermediates NA Dna2 Bard1 Brca1 Rad51 Rad51c Brip1 Rad51b Exo1 Gen1 Wrn Blm Brca2 Eme1
Hemostasis NA Fga Serpinf2 Atp1b1 Sparc Slc16a3 Plaur Plat Gpc1 Tgfb1 F11 Fam49b Arrb2 Fgb Rapgef4 Proc Ahsg Wee1 Vav2 Serping1 Esam Kif2c Syk Thbd Kif11 Igf1 Tfpi Rad51c Serpinb2 Lgals3bp Dock11 Prkcg Timp3 Racgap1 Col1a2 Rad51b Pde5a Nos3 Dock8 F7 Serpine2 Kif4 Tek Ehd3 Col1a1 Cav1 Prkch Fgg Serpind1 Serpina5 Igf2 Cd74 Ctsw Irf1 Fcer1g Pde9a Plek Rapgef3 Lcp2 P2rx7 Pde10a Kif15 Kif23 Csf2rb2 Gnai1 Zfpm1 Slc7a8 Spp2 Kif5a Il2rg Lck Rac2 Hist2h3c1 Gng2 Pla2g4a Cyb5r1 F8 Gata3 Cd63 Pf4 Serpina3c Kif3c P2ry12 Slc16a8 F2rl3 Ppia Vav3 Ttn Kif18a Pde3a Csf2 Prkcb
Resolution of D-Loop Structures NA Dna2 Bard1 Brca1 Rad51 Rad51c Brip1 Rad51b Exo1 Gen1 Wrn Blm Brca2 Eme1
Common Pathway of Fibrin Clot Formation NA Fga Fgb Proc Thbd Serpine2 Fgg Serpind1 Serpina5 F8 Pf4
Kinesins NA Kif2c Kif11 Racgap1 Kif4 Kif15 Kif23 Kif5a Kif3c Kif18a
Xenobiotics NA Cyp2c50 Cyp2c37 Cyp2c54 Cyp2e1 Cyp2c29 Cyp2f2 Cyp2c38 Cyp2b9 Cyp2c55 Arnt2 Cyp2a4
Resolution of D-loop Structures through Synthesis-Dependent Strand Annealing (SDSA) NA Dna2 Bard1 Brca1 Rad51 Rad51c Brip1 Rad51b Exo1 Wrn Blm Brca2
Initial triggering of complement NA Crp C2 C4b C3 C1qc C1qa C1qb Cfp
Integrin cell surface interactions NA Fga Icam1 Itgb6 Spp1 Fgb Col5a2 Itga2 Tnc Col18a1 Col1a2 Itga9 Col1a1 Fgg Col7a1 Icam2 Fbn1 Itga8 Col8a1 Col4a4
CRMPs in Sema3A signaling NA Dpysl3 Dpysl2 Cdk5r1 Dpysl5 Plxna4 Sema3a Crmp1 Plxna3
Phase 1 - Functionalization of compounds NA Adh1 Cyp4a14 Cyp2c50 Cyp2c37 Cyp4a10 Maob Cyp2c54 Cyp2e1 Cyp4b1 Cyp2c29 Cyp39a1 Cyp2f2 Cyp3a25 Nr1h4 Marc1 Fmo2 Aoc2 Aldh1b1 Cyp2c38 Cmbl Cyp27b1 Adh7 Aoc3 Cyp2b9 Cyp2c55 Fdx1l Arnt2 Cyp2a4
Meiotic Recombination NA Brca1 Rad51 Hist1h2bc Hist1h2bg Blm Brca2 Hist2h2aa2 Hist2h2aa1 Hist1h2bh Hist1h2bj Hist1h2bl Hist1h2bp Hist1h2bk Hist1h2ba H2afv Hist3h2a Hist1h2bn Hist1h2bf Hist1h2bm Hist2h3c1 Hist1h2bb Prdm9 Msh4 Hist2h2ac Hist1h4h
具体命令如下
#读取基因集文件
geneSets <- getGmt("test.geneset")
#读取表达量文件并去除重复
mydata <- read.table(file = "all.genes.fpkm.xls",header=T)
name=mydata[,1]
index <- duplicated(mydata[,1])
fildup=mydata[!index,]
exp=fildup[,-1]
row.names(exp)=name
#将数据框转换成矩阵
mydata= as.matrix(exp)
#使用gsva方法进行分析,默认mx.diff=TRUE,min.sz=1,max.zs=Inf,这里是设置最小值和最大值
res_es <- gsva(mydata, geneSets, min.sz=10, max.sz=500, verbose=FALSE, parallel.sz=1)
pheatmap(res_es)
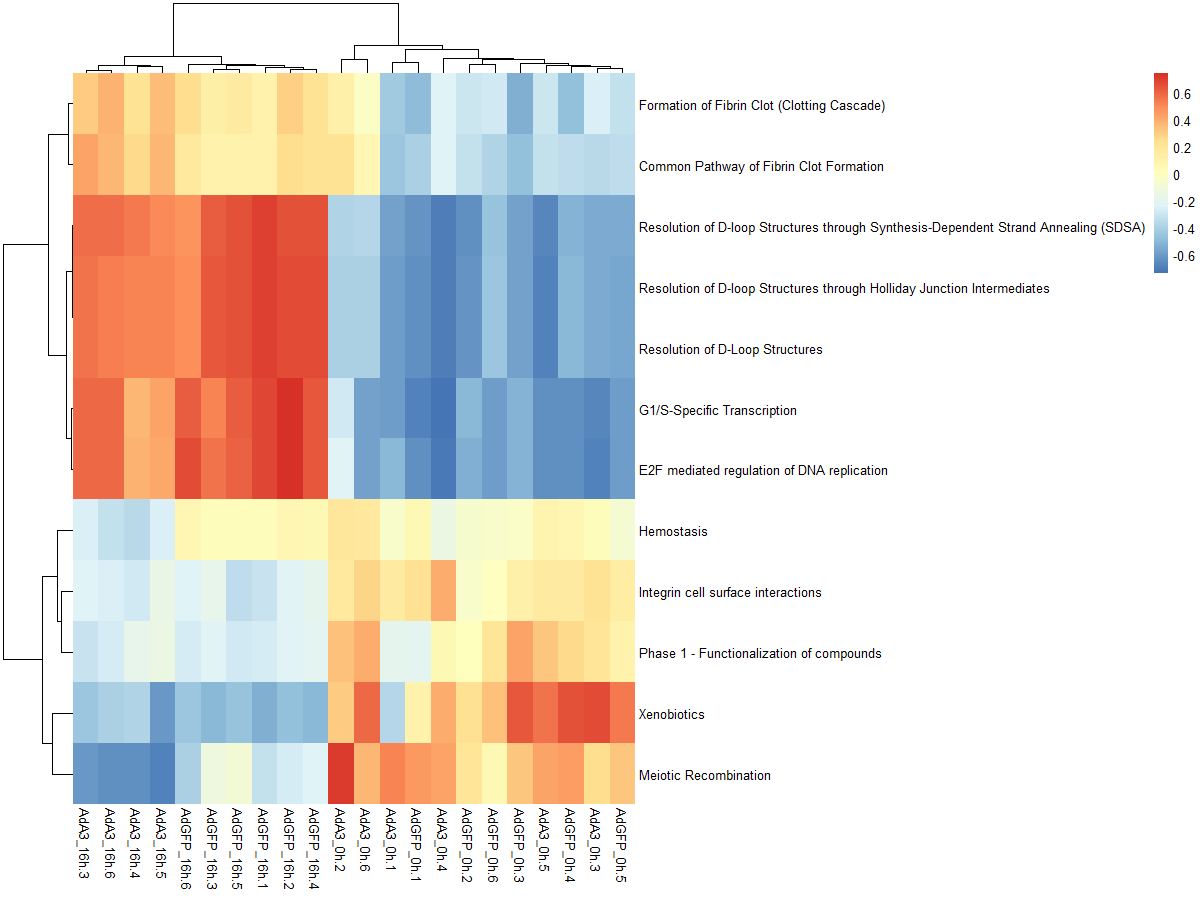
#mx.diff=FALSE es值是一个双峰的分布
es.max <- gsva(mydata, geneSets, mx.diff=FALSE, verbose=FALSE, parallel.sz=1)
pheatmap(es.max)
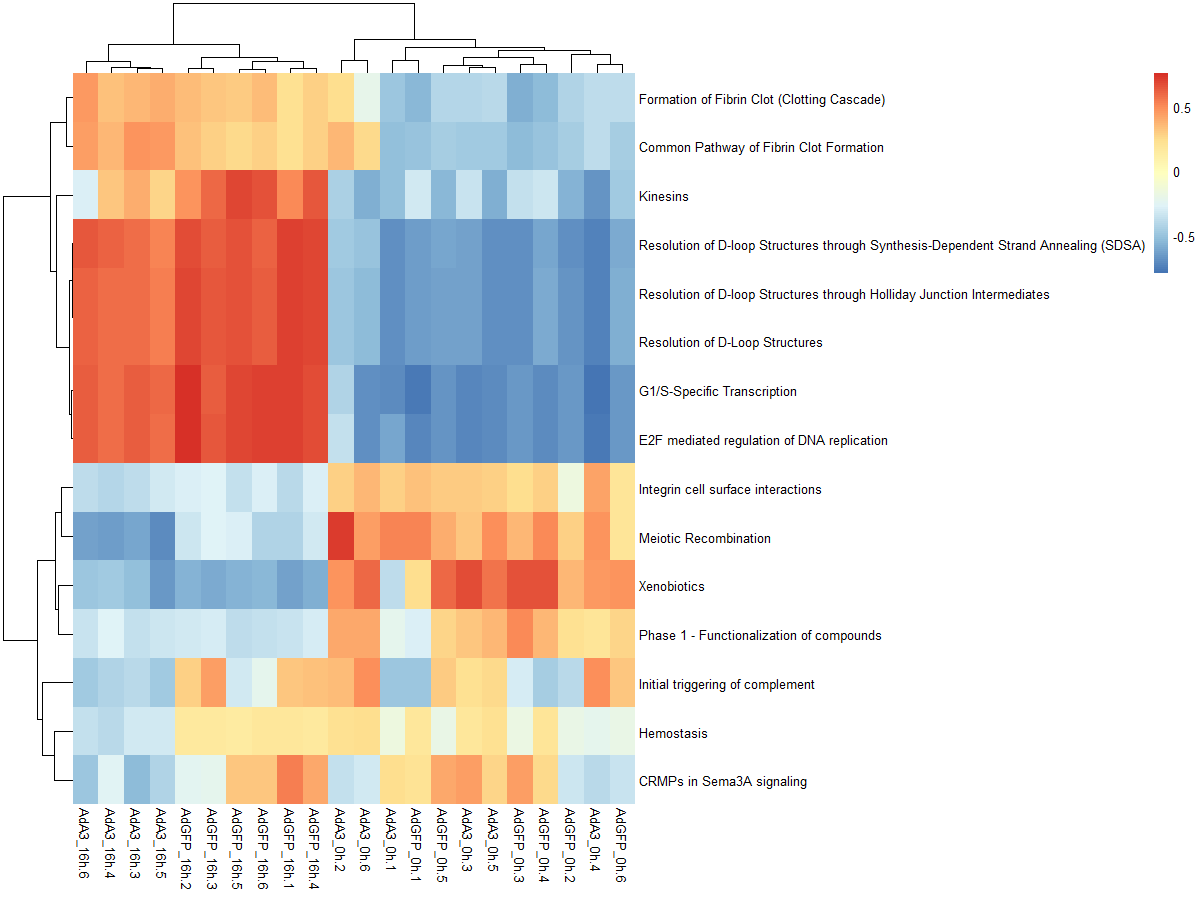
#mx.diff=TURE es值是一个近似正态分布
es.dif <- gsva(mydata, geneSets, mx.diff=TRUE, verbose=FALSE, parallel.sz=1)
pheatmap(es.dif)
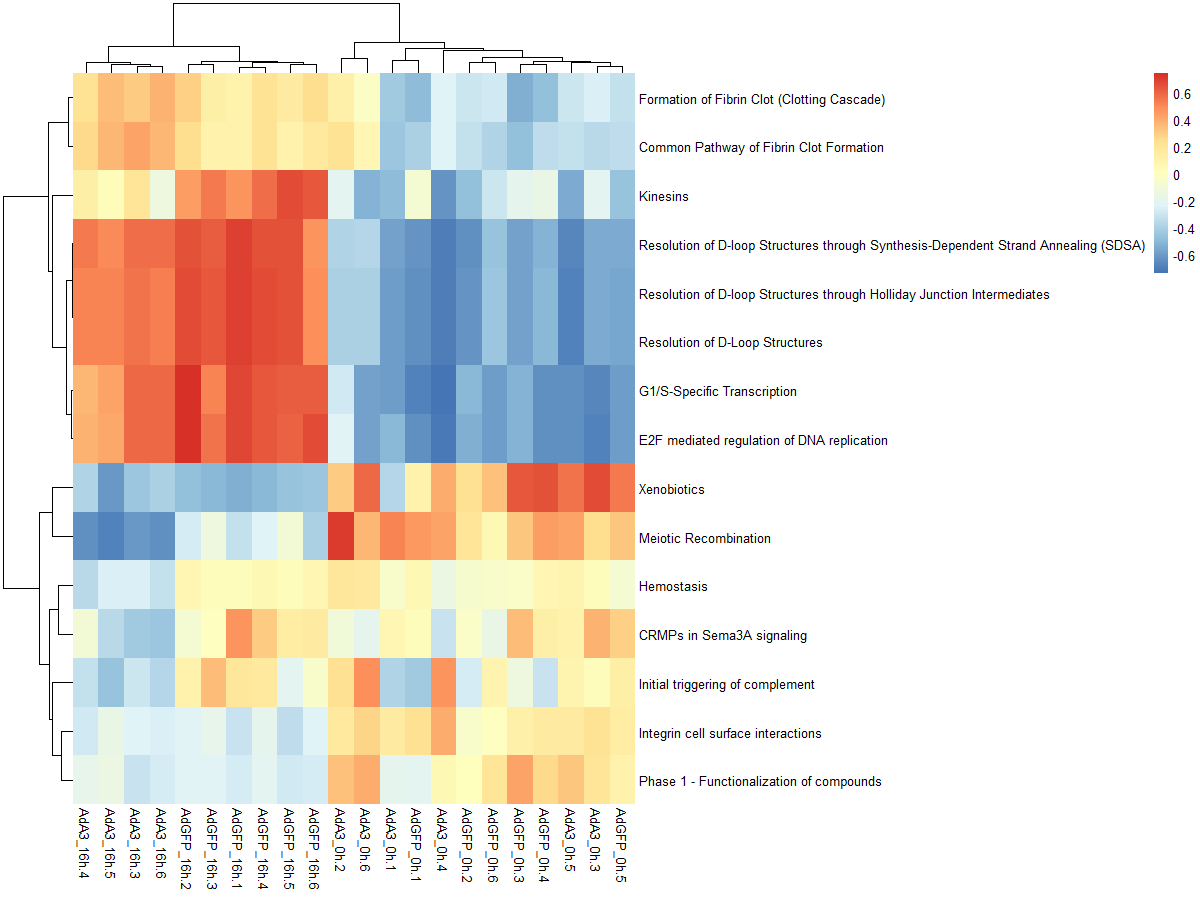
#可以看一下两种不同分布的效果,前者是高斯分布,后者是二项分布
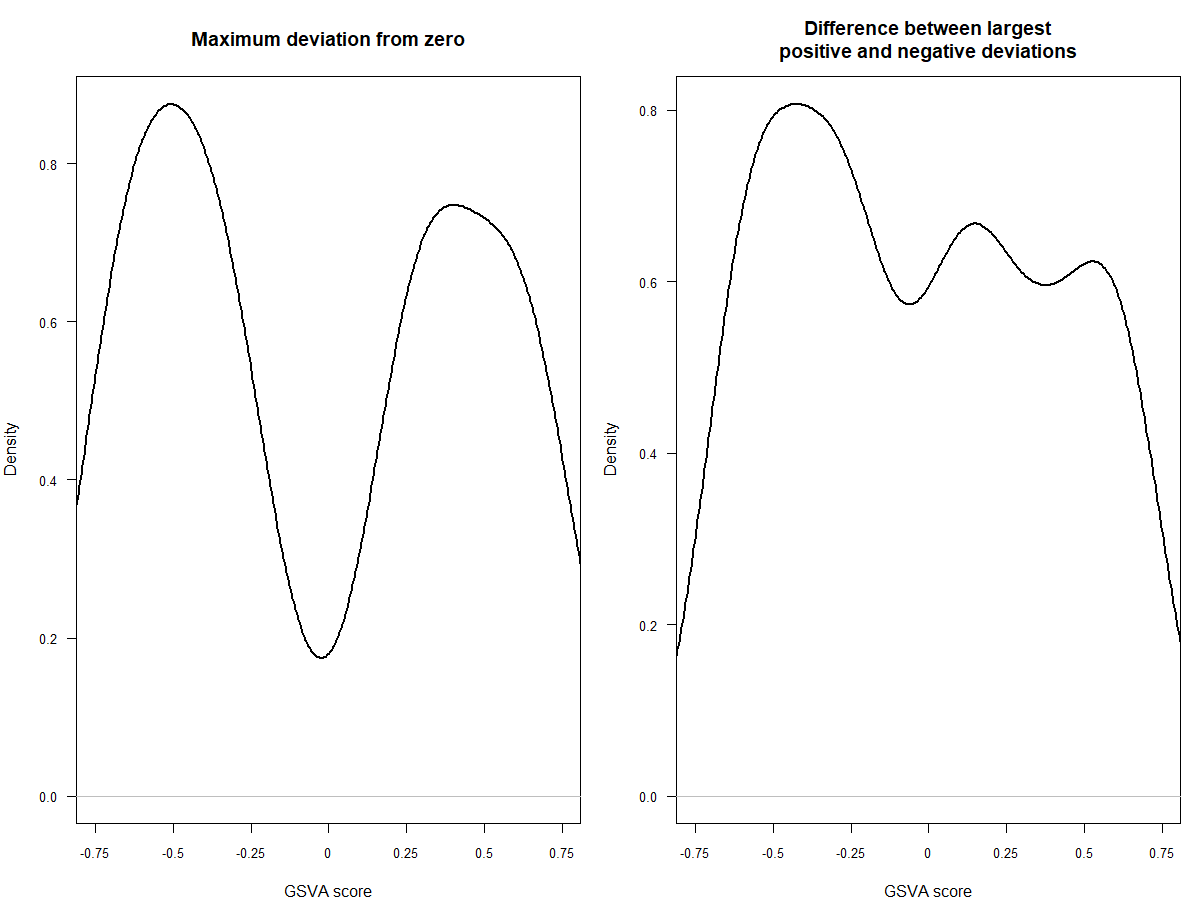
par(mfrow=c(1,2), mar=c(4, 4, 4, 1))
plot(density(as.vector(es.max)), main="Maximum deviation from zero",xlab="GSVA score", lwd=2, las=1, xaxt="n", xlim=c(-0.75, 0.75), cex.axis=0.8)
axis(1, at=seq(-0.75, 0.75, by=0.25), labels=seq(-0.75, 0.75, by=0.25), cex.axis=0.8)
plot(density(as.vector(es.dif)), main="Difference between largest\npositive and negative deviations",xlab="GSVA score", lwd=2, las=1, xaxt="n", xlim=c(-0.75, 0.75), cex.axis=0.8)
axis(1, at=seq(-0.75, 0.75, by=0.25), labels=seq(-0.75, 0.75, by=0.25), cex.axis=0.8)
- limma设置分组的方法
group_list <- factor(c(rep("control",2), rep("siSUZ12",2)))
design <- model.matrix(~group_list)
colnames(design) <- levels(group_list)
rownames(design) <- colnames(counts)
#设置分组
col = names(exp)
sample=col[1:10]
group=c(rep('control',6),rep('treat',4))
phno=data.frame(sample,group)
Group<-factor(phno$group,levels=levels(phno$group))
design<-model.matrix(~0+Group)
colnames(design) <- c("control", "treat")
#获取需要进行差异分析的分组
res=es.max[,1:10]
#定义阈值
logFCcutoff <- log2(1.5)
adjPvalueCutoff <- 0.001
#进行差异分析
fit <- lmFit(res, design)
fit <- eBayes(fit)
allGeneSets <- topTable(fit, coef="treat", number=Inf)
DEgeneSets <- topTable(fit, coef="treat", number=Inf,p.value=adjPvalueCutoff, adjust="BH")
res <- decideTests(fit, p.value=adjPvalueCutoff)
#summary(res)
#画火山图
DEgeneSets$significant="no"
DEgeneSets$significant=ifelse(DEgeneSets$logFC>0|DEgeneSets$logFC<0,"up","down")
ggplot(DEgeneSets,aes(logFC,-1*log10(adj.P.Val)))+geom_point(aes(color =significant)) + xlim(-4,4) + ylim(0,30)+labs(title="Volcanoplot",x="log[2](FC)", y="-log[10](FDR)")+scale_color_manual(values =c("#00ba38","#619cff","#f8766d"))+geom_hline(yintercept=1.3,linetype=4)+geom_vline(xintercept=c(-1,1),linetype=4)
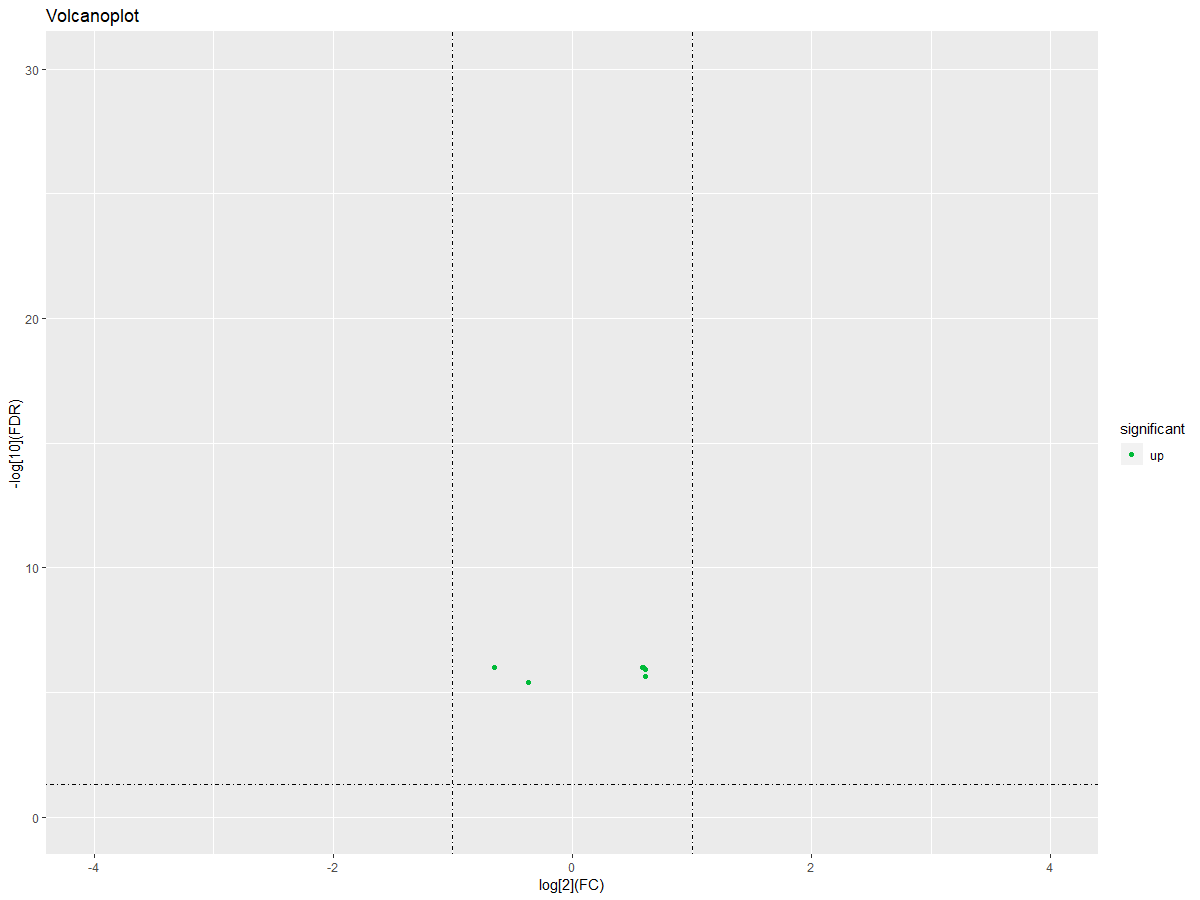
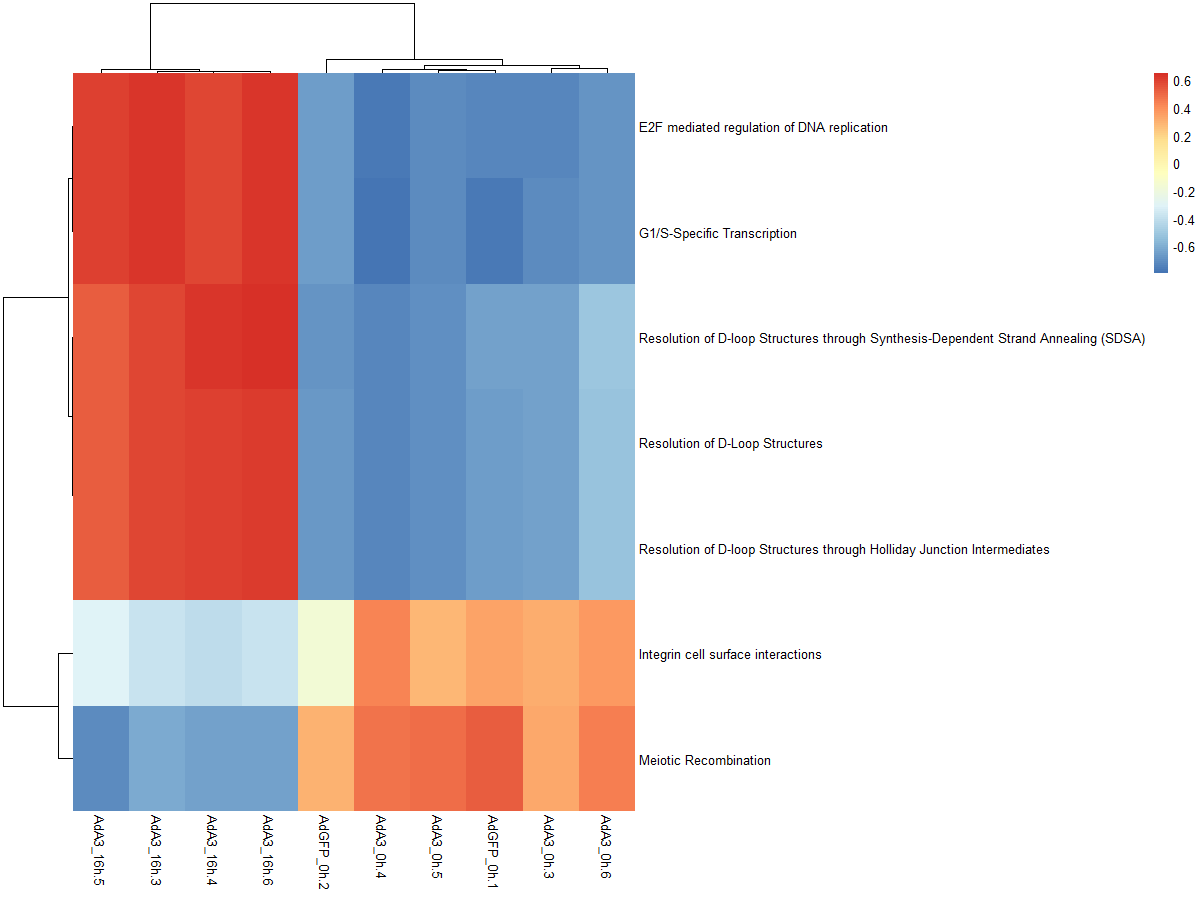
#获取差异基因集的表达量
DEgeneSetspkm = merge(DEgeneSets,es.max,by=0,all.x=TRUE)[,c(1,11:20)]
degsetsp=DEgeneSetspkm[,-1]
name=DEgeneSetspkm[,1]
row.names(degsetsp)=name
pheatmap(degsetsp)
GSVA的使用的更多相关文章
- 根据GSVA结果绘制不同组的趋势图
首先需要将GSVA的矩阵结果转换成如下格式: 然后使用如下代码进行作图 infile <- "draw_pre_violin_heatmap.txt" data <- ...
- 深入解析SQL Server并行执行原理及实践(下)
谈完并行执行的原理,咱们再来谈谈优化,到底并行执行能给我们带来哪些好处,我们又应该注意什么呢,下面展开. Amdahl’s Law 再谈并行优化前我想有必要谈谈阿姆达尔定律,可惜老爷子去年已经驾鹤先 ...
- R 动态定义变量名 assign
rm(list=ls()) library(GSVA) library(GSEABase) library(GSVAdata) library(msigdbr) library(org.Hs.eg.d ...
随机推荐
- SignalR简单实用_转自:https://www.cnblogs.com/humble/p/3851205.html
一.指定通信方式 建立一个通讯方式需要一定的时间和客户机/服务器资源.如果客户机的功能是已知的,那么通信方式在客户端连接开始的时候就可以指定.下面的代码片段演示了使用AJAX长轮询方式来启动一个连接, ...
- CPU突然飙升到300%,Dubbo活动线程数直接飙到1000
转:https://mp.weixin.qq.com/s/-lSiVDfqYrKk_w-xitYBhA 背景:新功能开发测试完成后,准备发布上线,当发布完第三台机器时,监控显示其中一台机器CPU突然飙 ...
- 大数据学习之路之HBASE
Hadoop之HBASE 一.HBASE简介 HBase是一个开源的.分布式的,多版本的,面向列的,半结构化的NoSql数据库,提供高性能的随机读写结构化数据的能力.它可以直接使用本地文件系统,也可以 ...
- Gremlin入门
Gremlin入门 一.Gremlin简介 Gremlin是Apache ThinkerPop框架下的图遍历语言,Gremlin是一种函数式数据流语言,可以使用户使用简洁的方式表述复杂的属性图的遍历或 ...
- SpringContextHolder 作用
以静态变量保存Spring ApplicationContext, 可在任何代码任何地方任何时候取出ApplicaitonContext. 使用方式.在启动类里添加Bean package com.y ...
- Mybatis笔记(二)
目录 MyBatis 逆向工程 MyBatis Generator 使用 分页插件 1.下载分页插件 2.配置分页插件 3.使用分页插件 SSM整合(spring与springMVC) 1.创建web ...
- git clone速度太慢的解决办法
最近发现使用git clone的速度比较慢,于是找到了办法分享给大家: 思路: git clone特别慢是因为github.global.ssl.fastly.net域名被限制了. 只要找到这个域名对 ...
- if ( ! defined('BASEPATH')) exit('No direct script access allowed')的作用
在看源代码时,发现codeigniter框架的控制器中,总是加上这样一段话: if(!defined('BASEPATH'))exit('No direct script access allowed ...
- 降维算法整理--- PCA、KPCA、LDA、MDS、LLE 等
转自github: https://github.com/heucoder/dimensionality_reduction_alo_codes 网上关于各种降维算法的资料参差不齐,同时大部分不提供源 ...
- linux 查看gpu信息