topGO
前面我们讲过GO.db这个包,现在接着延伸topGO包,该包是用来协助GO富集分析
1)安装
if("topGO" %in% rownames(installed.packages()) == FALSE) {source("http://bioconductor.org/biocLite.R");biocLite("topGO")}
suppressMessages(library(topGO))
ls("package:topGO")
2)使用方法
该包主要有三个使用步骤:
2.1、Data preparation:准备数据集,用于构建 topGOdata.对象。
2.1.1、包括gene标识符(List of genes identifiers)及相应的分值gene scores(例如p值等)
2.1.2、差异表达基因list或经一定标准按照分值筛选的基因集用于后续分析(list of differentially expressed genes or a criteria for selecting genes
based on their scores);
2.1.3、identifier和GO term间的map,即GOterm表:(gene-to-GO annotations ) ####例如测试文件中的geneid2go.map
2.1.4、GO的层级结构,由GO.db提供,目前这个包只支持GO.db提供的结构
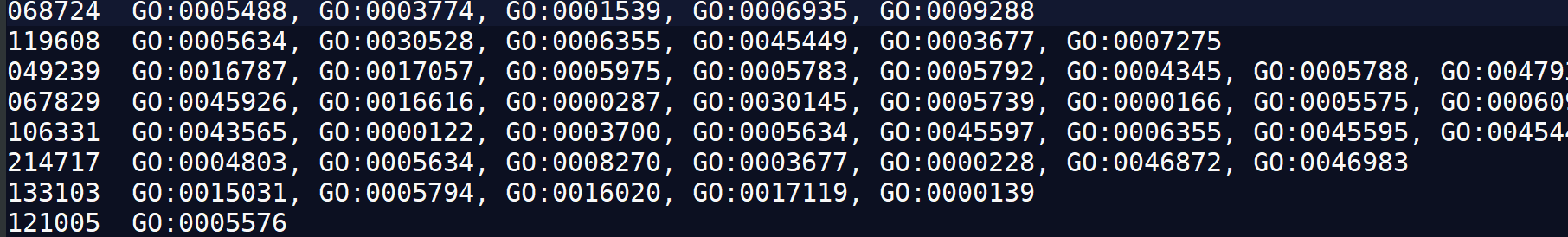
goterm表示例(gene-to-GO annotations ):
2.2、Running enrichment tests:进行富集分析,用任何可行的混合统计测试和方法来处理 GO拓扑结构(GO topology)
2.3、Analysis results:用 summary functions 和 visualisation tools对第二步进行统计和可视化
3)简单示例(guide)
3.1.1、准备输入文件
library(ALL)
data(ALL)
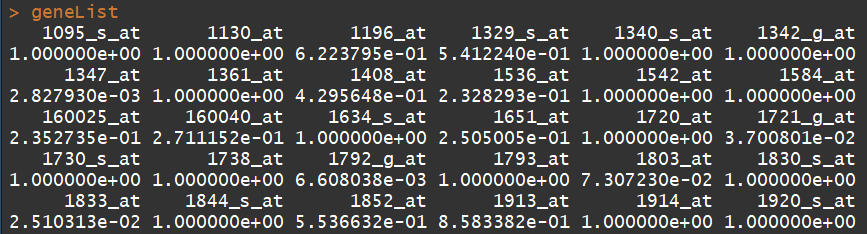
data(geneList) ##文件1:基因list,
affyLib <- paste(annotation(ALL), "db", sep = ".") #####"hgu95av2.db"
library(package = affyLib, character.only = TRUE) ########GO term表
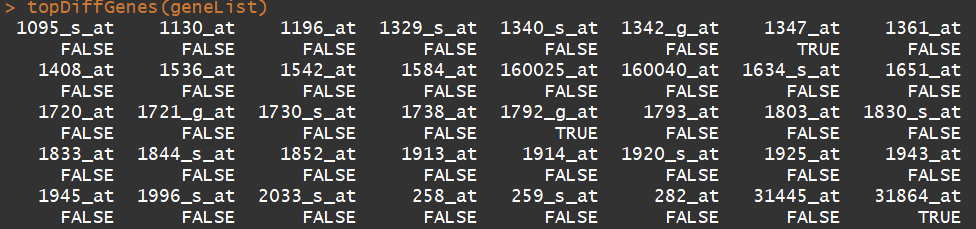
sum(topDiffGenes(geneList) ###选择差异基因集,
3.1.2、构建 topGOdata对象(核心步骤):
sampleGOdata <- new("topGOdata",
description = "Simple session", ##topGOdata的描述,可选
ontology = "BP", ##可指定要分析的GO term的类型,即BP、CC之类
allGenes = geneList, ##基因identifier的原始列表
geneSel = topDiffGenes, ##geneSelectionFun联合作用,筛选出后续参与分析的基因
nodeSize = 10, ##富集的GO term辖下基因的最小数目,这里选择10.即最少10个
annot = annFUN.db, ##提取gene-to-GO mappings 的对应关系
affyLib = affyLib)
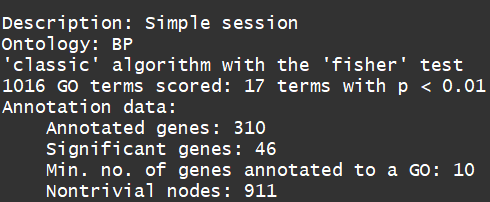
sampleGOdata

3.2 Performing the enrichment tests
有了topGOdata对象,接下来就可以用来进行富集分析。这里用两种检验方法:Fisher’s exact test (基于 gene counts)和Kolmogorov-Smirnov like test (computes enrichment based on gene scores)。
其中用runTest函数来进行这些检验,该函数含有3个参数:第一个是topGOdata对象、第二个是algorithm(用于指定处理 GO graph structured的方法)、第三个是statistic(用于指定检验方法)
resultFisher <- runTest(sampleGOdata, algorithm = "classic", statistic = "fisher") ##Fisher’s exact test
resultKS <- runTest(sampleGOdata, algorithm = "classic", statistic = "ks") #Kolmogorov-Smirnov test,classic method
resultKS.elim <- runTest(sampleGOdata, algorithm = "elim", statistic = "ks")#Kolmogorov-Smirnov test, elim method
3.3 Analysis of results
当富集检验结束后,我们就可以分析并解析结果。
runTest()这个函数用来分析显著富集的 GO terms及其相应的p值。
allRes <- GenTable(sampleGOdata, ##之前构建的topGOdata实例
classicFisher = resultFisher, ##生成GO graphde的方法
classicKS = resultKS, ##生成GO graphde的方法
elimKS = resultKS.elim, ##生成GO graphde的方法
orderBy = "elimKS",
ranksOf = "classicFisher",
topNodes = 10) ##这里显示前10个显著结果
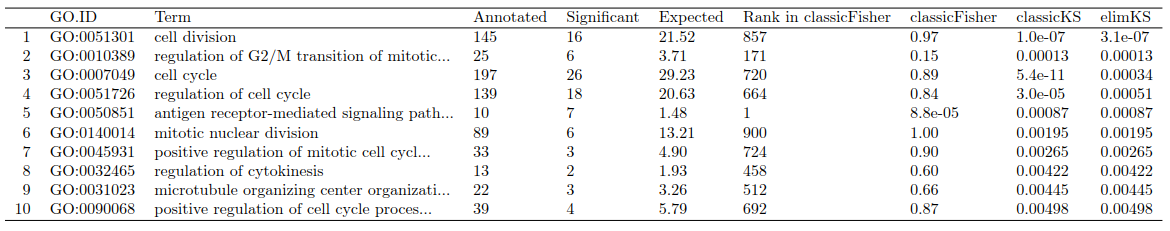
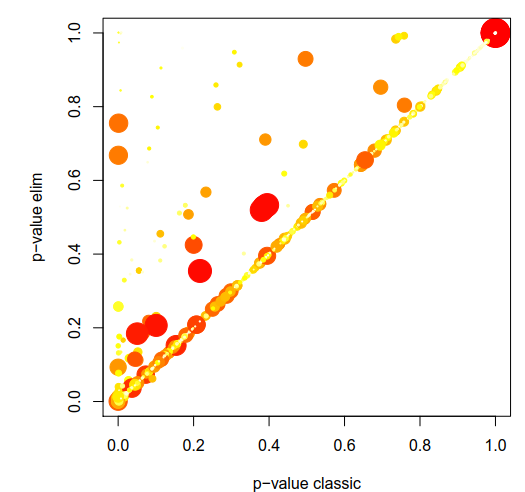
用 score()函数来测评topGO结果对象中 GO term的 p-values ,并用散点图来说明。
pValue.classic <- score(resultKS)
pValue.elim <- score(resultKS.elim)[names(pValue.classic)]
gstat <- termStat(sampleGOdata, names(pValue.classic))
gSize <- gstat$Annotated / max(gstat$Annotated) * 4
plot(pValue.classic, pValue.elim, xlab = "p-value classic", ylab = "p-value elim",pch = 19, cex = gSize)
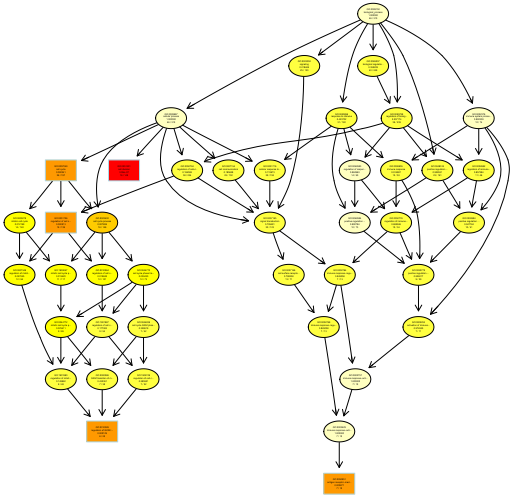
差看显著富集的GO terms在 GO graph中的分布.
showSigOfNodes(sampleGOdata, score(resultKS.elim), firstSigNodes = 5, useInfo = 'all')
4)实战
4.1 原始数据集的准备(上面的4个文件)
library(topGO)
library(ALL) ##准备数据集
data(ALL) ##文件1:原始数据集
BPterms <- ls(GOBPTerm)
MFterms <- ls(GOMFTerm)
CCterms <- ls(GOCCTerm)
head(BPterms)
head(MFterms)
head(CCterms) library(genefilter) ##对原始数据进行过滤
selProbes <- genefilter(ALL, filterfun(pOverA(0.20, log2(100)), function(x) (IQR(x) > 0.25)))#数据清洗
eset <- ALL[selProbes, ] ##数据清洗:这里去掉及其低表达的基因,及探针在每个样品中表达变化不大的的基因
myInterestingGenes <- sample(geneNames, length(geneNames) / 10) #文件二:经一定标准对p值等筛选获取感兴趣基因集用于后续分析
geneList <- factor(as.integer(geneNames %in% myInterestingGenes))
names(geneList) <- geneNames
str(geneList) geneID2GO <- readMappings(file = system.file("examples/geneid2go.map", package = "topGO"))##文件三:goterm的map文件
str(head(geneID2GO)) GO2geneID <- inverseList(geneID2GO) ###额外知识:用inverseList()函数实现gene-to-GOs与 GO-to-genes 之间的转换
str(head(GO2geneID)) ##
topGO的更多相关文章
- 10、差异基因topGO富集
参考:http://www.biotrainee.com/thread-558-1-1.html http://bioconductor.org/packages/3.7/bioc/ http://w ...
- R: 修改镜像、bioconductor安装及go基因富集分析
1.安装bioconductor及go分析涉及的相关包 source("http://bioconductor.org/biocLite.R") options(BioC_mirr ...
- 20155205 2016-2017-2 《Java程序设计》第6周学习总结
20155205 2016-2017-2 <Java程序设计>第6周学习总结 教材学习内容总结 第十章 在Java中,输入串流代表对象为Java.io.InputStream实例,输出串流 ...
- 20155318 2016-2017-2 《Java程序设计》第六周学习总结
20155318 2016-2017-2 <Java程序设计>第六周学习总结 教材学习内容总结 学习目标 理解流与IO 理解InputStream/OutPutStream的继承架构 理解 ...
- 20155328 2016-2017-2 《Java程序设计》第六周 学习总结
20155328 2016-2017-2 <Java程序设计>第6周学习总结 教材学习内容总结 根据不同的分类标准,IO可分为:输入/输出流:字节/字符流:节点/处理流. 在不使用Inpu ...
- 20155339 2016-2017-2 《Java程序设计》第6周学习总结
20155339 2016-2017-2 <Java程序设计>第6周学习总结 教材学习内容总结 第十章 串流设计 Java将输入/输出抽象化为串流,数据有来源及目的地,衔接两者的是串流对象 ...
- 20155224 2016-2017-2 《Java程序设计》第6周学习总结
20155224 2016-2017-2 <Java程序设计>第6周学习总结 教材学习内容总结 Thread线程: 定义某线程后,要有 xxx.stard(); Thread.sleep( ...
- 20155237 2016-2017-2 《Java程序设计》第6周学习总结
20155237 2016-2017-2 <Java程序设计>第6周学习总结 教材学习内容总结 第十章 输入与输出 InputStream与OutputStream 流(Stream)是对 ...
- 20155229 2016-2017-2 《Java程序设计》第六周学习总结
20155229 2016-2017-2 <Java程序设计>第六周学习总结 教材学习内容总结 第十章 Java中,输入串流代表对象为java.io.InputStream,输出串流代表对 ...
随机推荐
- Django 实现CRM 问卷调查功能组件
目录结构: 母版 {% load staticfiles %} <!DOCTYPE html> <html lang="zh-CN"> <head&g ...
- C#实现不安装Oracle客户端访问远程服务器数据
概述: C#通过使用ADO的方式在未安装Oracle数据库的前提下,客户端程序远程访问服务器,会出现:“System.Data.OracleClient 需要 Oracle 客户端软件 8.1.7 或 ...
- wxWidgets:处理wxEVT_PAINT
我们仍然以继承于wxFrame的MyFrame作为例子. MyFrame.h: class MyFrame : public wxFrame { ...... private: ...... void ...
- SpringAOP基础 - 静态代理设计模式
代理模式在实现过程中,要创建一个接口(社交技巧-接口),代理类(经纪人 - 类)和真实类(范冰冰 - 类)同时实现这个接口. 举个例子: 我们想要找范冰冰吃饭,但是呢,她是大明星,不可能轻易见我们,我 ...
- 用Keras搭建神经网络 简单模版(一)——Regressor 回归
首先需要下载Keras,可以看到我用的是TensorFlow 的backend 自己构建虚拟数据,x是-1到1之间的数,y为0.5*x+2,可视化出来 # -*- coding: utf-8 -*- ...
- 捷通华声TTS在Aster+中的安装过程
1)挂载TTS光碟 2)安装如下5个rpm软件包 [asterisk@TTS78:/mnt]$ls *.rpmjTTS-5.0.1.0-3.i386.rpm VocLib_Xi ...
- Android RIL概述
前言 Android作为一个通用的移动平台,其首要的功能就是通话.短信以及上网等通信功能.那么,从系统的角度来看,Android究竟是怎么实现与网络的交互的了? 这篇文章里,就来看一看Android中 ...
- 第10课 C++中的新成员
1. 动态内存分配 (1)C++通过new关键字进行动态内存申请,是以类型为单位来申请空间大小的 (2)delete关键字用于内存释放 ▲注意释放数组时要加[],否则只释放这个数组中的第1个元素. [ ...
- Flex知识
转载:http://www.cnblogs.com/xia520pi/archive/2011/12/11/2283851.html
- tomcat启动项目 端口占用
转自:https://blog.csdn.net/u010427935/article/details/77297529 有时候电脑比较卡,项目比较大的情况下,eclipse没有完全停止tomcat的 ...