利用ONT测序检测真核生物全基因组甲基化状态
摘要
甲基化在真核生物基因组序列中广泛存在,其中5mC最为普遍,在真核生物基因组中也有发现6mA。捕获基因组中的甲基化状态的常用技术是全基因组甲基化测序(WGBS)和简化甲基化测序(RRBS),而随着第三代测序技术的完善,ONT单分子纳米孔测序可以从单分子的角度来检出甲基化的胞嘧啶和腺嘌呤电流的变化,从而实现由基因组中的一段序列中检出5mC和6mA,然而精确地从单碱基级别检出5mC和6mA扔具有挑战。本文利用第三代ONT测序技术获得的序列及其电信号来检出真核生物全基因组范围的5mC和6mA甲基化状态。
背景
DNA甲基化主要发生在脱氧核糖核苷酸的第五位的胞嘧啶和第六位的腺嘌呤,前者普遍存在于真核生物,后者在原核生物中广泛存在,也有研究报道6mA存在于真核生物。这样的甲基化状态在ONT测序仪捕捉到的电流信号中,不仅单碱基的电流会发生改变,而且其上下文的一段基因组序列也会发生改变。基于此,一些生物信息学软件先后被开发出来针对于这两种甲基化的检出有各自的优缺点。有研究指出在真核基因组中检出5mC和6mA准确度较高的软件分别为nanopolish[1]和tombo[2]。
利用nanopolish检出真核生物基因组中5mC的甲基化位置
材料和方法
利用minION平台对目标生物血液提取的DNA不打断建库并进行全基因组测序,获得12G序列及其电信号文件。安装nanopolish(v0.13.2)。
步骤
- 建立索引
nanopolish index -d fast5_files/ output.fastq
- 比对
minimap2 -a -x map-ont reference.fasta output.fastq | samtools sort -T tmp -o output.sorted.bam
samtools index output.sorted.bam
- Calling methylation
nanopolish call-methylation --progress -q cpg -t NCPU --verbose -r reads.fastq -b output.sorted.bam -g reference_genome.fasta > nanopolish_call_methylation.tsv
- 筛选高置信度的甲基化和未甲基化位点
calculate_methylation_frequency.py[3]
nanopore-methylation-utilities/mtsv2bedGraph.py[4]
scripts/calculate_methylation_frequency.py -c 2 methylation_calls.tsv > methylation_frequency.tsv
# or
python nanopore-methylation-utilities/parseMethylbed.py frequency -i methylation_calls.tsv -o methylation_calls_freq.tsv --verbose -m cpg -u 2 -l -2
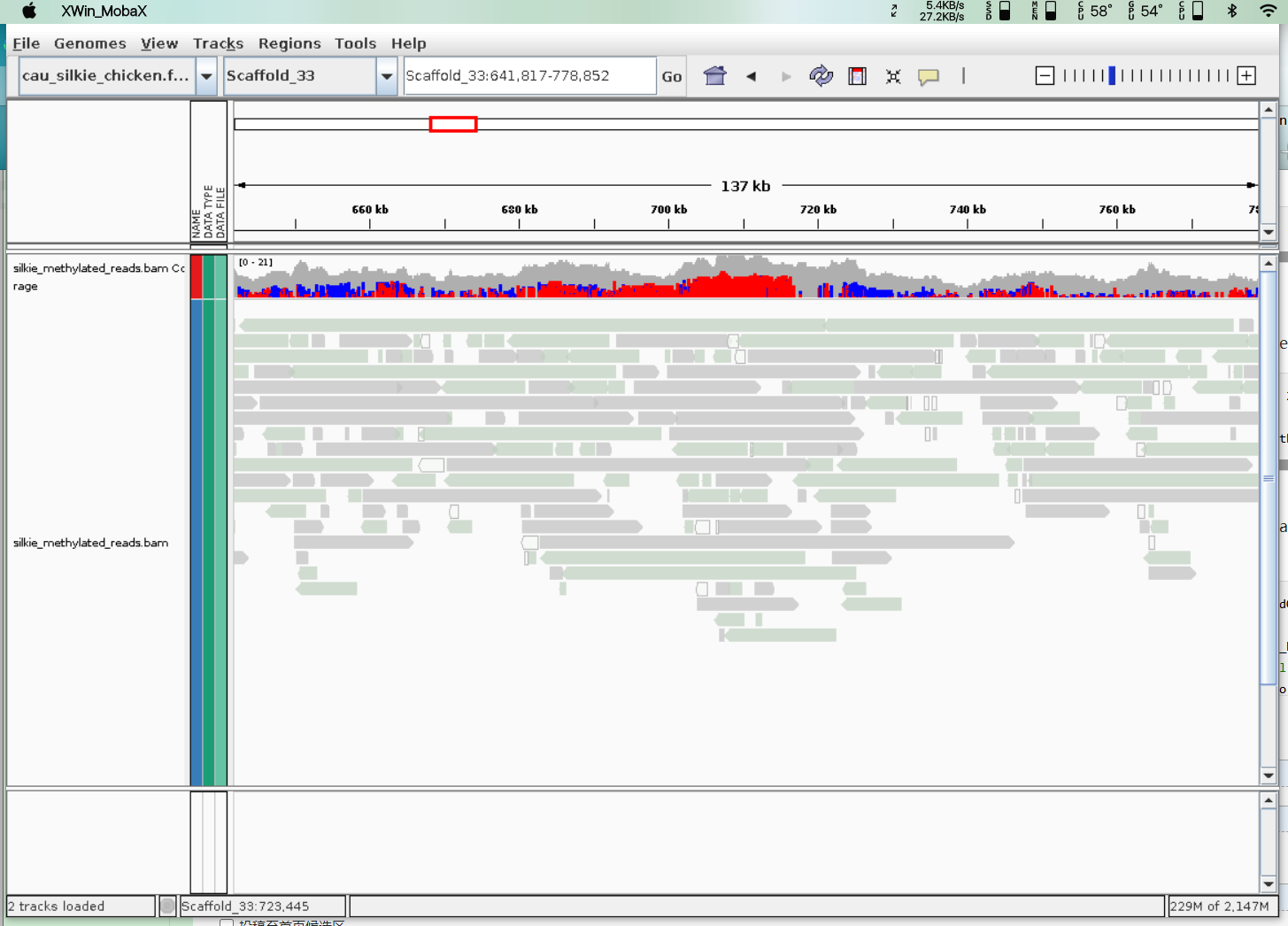
- IGV可视化或UCSC可视化
IGV可视化[5]
python nanopore-methylation-utilities/mtsv2bedGraph.py --verbose -c 2 -i methylation_calls.tsv -q cpg -g reference_genome.fasta | sort -k1,1 -k2,2n | bgzip > methylation_calls.bed.gz
tabix -p methylation_calls.bed.gz
python nanopore-methylation-utilities/convert_bam_for_methylation.py -t 100 --verbose --remove_poor -c methylation_calls.bed.gz -f reference_genome.fasta -b output.sorted.bam | samtools sort -o methylation_calls.bed.remove_no_or_poor_methylation_reads.bam
# if all reads' coverage was needed, remap all reads to reference to find out
samtools index methylation_calls.bed.remove_no_or_poor_methylation_reads.bam
# now bam file can be loaded to igv via their bisulfite mode to see methylation sites and unmethylation sites.
利用ONT测序检测真核生物全基因组甲基化状态的更多相关文章
- 全基因组测序 Whole Genome Sequencing
全基因组测序 Whole Genome Sequencing 全基因组测序(Whole Genome Sequencing,WGS)是利用高通量测序平台对一种生物的基因组中的全部基因进行测序,测定其 ...
- cfDNA(circulating cell free DNA)全基因组测序
参考资料: [cfDNA专题]cell-free DNA在非肿瘤疾病中的临床价值(好) ctDNA, cfDNA和CTCs有什么区别吗? cfDNA你懂多少? 新发现 | 基因是否表达,做个cfDNA ...
- 全基因组测序 从头测序(de novo sequencing) 重测序(re-sequencing)
全基因组测序 全基因组测序分为从头测序(de novo sequencing)和重测序(re-sequencing). 从头测序(de novo)不需要任何参考基因组信息即可对某个物种的基因组进行测序 ...
- PacBio全基因组测序和组装
PacBio公司的业务范围也就5个(官网): Whole Genome Sequencing Targeted Sequencing Complex Populations RNA Sequencin ...
- WGS 全基因组测序数据分析
1. DNA测序技术 https://www.jianshu.com/p/6122cecec54a 2.FASTA和FASTQ文件格式 https://www.jianshu.com/p/50ff30 ...
- GWAS | 全基因组关联分析 | Linkage disequilibrium (LD)连锁不平衡 | 曼哈顿图 Manhattan_plot | QQ_plot | haplotype phasing
现在GWAS已经属于比较古老的技术了,主要是碰到严重的瓶颈了,单纯的snp与表现的关联已经不够,需要具体的生物学解释,这些snp是如何具体导致疾病的发生的. 而且,大多数病找到的都不是个别显著的snp ...
- 如何鉴定全基因组加倍事件(WGD)
目前鉴定全基因组加倍(whole-genome duplication events)有3种 通过染色体共线性(synteny) 方法是比较两个基因组的序列,并将同源序列的位置绘制成点状图,如果能在点 ...
- 【GWAS文献解读】疟原虫青蒿素抗药性的全基因组关联分析
英文名:Genetic architecture of artemisinin-resistant Plasmodium falciparum 中文名:疟原虫青蒿素抗药性的全基因组关联分析 期刊:Na ...
- Genome-wide Complex Trait Analysis(GCTA)-全基因组复杂性状分析
GCTA(全基因组复杂性状分析)工具开发目的是针对复杂性状的全基因组关联分析,评估SNP解释的表型方差所占的比例(该网站地址:http://cnsgenomics.com/software/gcta/ ...
随机推荐
- ceph总结复习
一.ceph概念 Ceph是一种为优秀的性能.可靠性和可扩展性而设计的统一的.分布式文件系统.ceph 的统一体现在可以提供文件系统.块存储和对象存储,分布式体现在可以动态扩展. 什么是块存储/对象存 ...
- 完全理解Python 迭代对象、迭代器、生成器
在了解Python的数据结构时,容器(container).可迭代对象(iterable).迭代器(iterator).生成器(generator).列表/集合/字典推导式(list,set,dict ...
- Google I/O 2021 Android精华内容
Google I/O 2021结束了, 都有什么精彩内容呢? Android部分的Playlist附上: Android & Play at Google I/O 2021 Developer ...
- skynet debug console 使用
预读 关于如何使用 skynet 可以参考 wiki 文档 更多实战内容见 https://www.lanqiao.cn/courses/2770 优惠码:2CZ2UA5u 环境测试搭建 使用示例代码 ...
- 浅谈:Redis持久化机制(一)RDB篇
浅谈:Redis持久化机制(一)RDB篇 众所周知,redis是一款性能极高,基于内存的键值对NoSql数据库,官方显示,它的读效率可达到11万次每秒,写效率能达到8万次每秒,因为它基于内存以及存 ...
- App元素定位三种方法
来自博客: http://testingpai.com/article/1595507262082 以下方法操作前必须确保有手机设备连入电脑,检测是否有手机连入命令 adb devices 第一种:A ...
- ONNX MLIR方法
ONNX MLIR方法 MLIR中的开放式神经网络交换实现. Prerequisites gcc >= 6.4 libprotoc >= 3.11.0 cmake >= 3.15.4 ...
- 4D雷达成像技术
4D雷达成像技术 当我们谈及3D捕捉时,总是先想到光学传感器.当我们讨论在第四维度(时间)讨论视觉数据时,倾向于考虑场景数据调度.这些是我们多年来关注激光雷达(LiDAR)和摄影测量,以及用户针对缓慢 ...
- SpringBoot基础系列之自定义配置源使用姿势实例演示
[SpringBoot基础系列]自定义配置源的使用姿势介绍 前面一篇博文介绍了一个@Value的一些知识点,其中提了一个点,@Value对应的配置,除了是配置文件中之外,可以从其他的数据源中获取么,如 ...
- 【NX二次开发】Block UI 集列表
属性说明 属性 类型 描述 常规 BlockID String 控件ID Enable Logical 是否可操作 Group ...