单细胞测序最好的教程(十四)测序原始数据公开至NCBI数据库
作者按
国内对于单细胞测序相关的中文教程确实不够全面,当然NCBI官网给的上传教程也比较详细了,所以变成了会者不难。本教程你现在可能用不上,但是你如果做单细胞测序,那么未来你一定会用上,建议收藏。
在这里,我们将演示如何将测序文件完整上传到NCBI上。本教程首发于单细胞最好的中文教程,未经授权许可,禁止转载。
全文字数|预计阅读时间: 3500|5min
——星夜老师
1. 注册NCBI账户
我们首先打开SRA的上传官网:https://submit.ncbi.nlm.nih.gov/subs/sra/,注册一个账户
注意
不能使用163,qq邮箱之类的,可能会收不到邮件
2. 新建SRA提交
2.1 选择Biosample
我们首先需要准备biosample,这个是需要ncbi官网审核的,每一个biosample代表了一个真实的样本。
2.2 填写信息
一般来说,我们可以指定一个数据的释放日期,我估计一年的时间论文应该能发出去,所以设置到了明年同一时间。
下拉选择人类资源Human
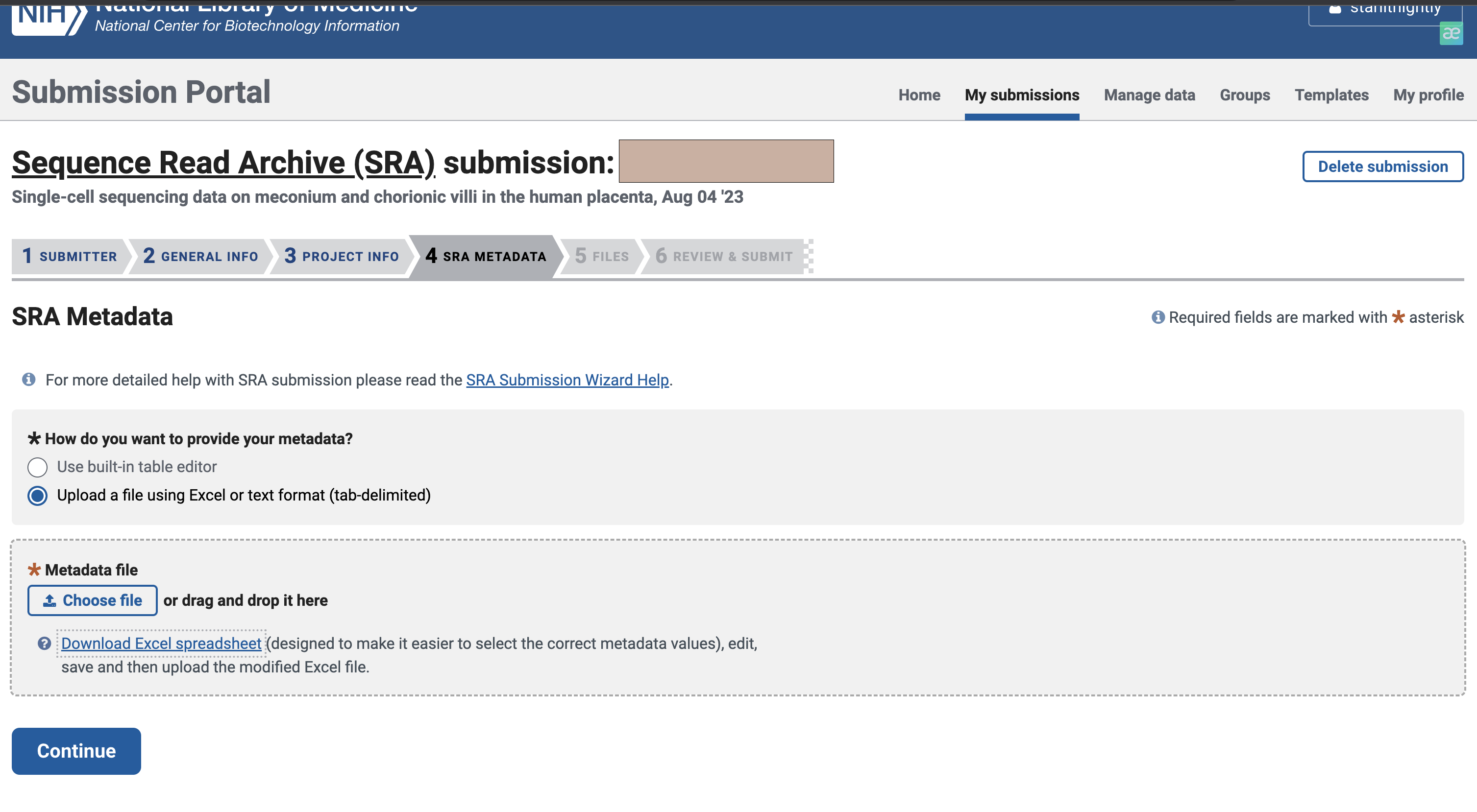
在这里,我们选择文件上传的方式进行填写
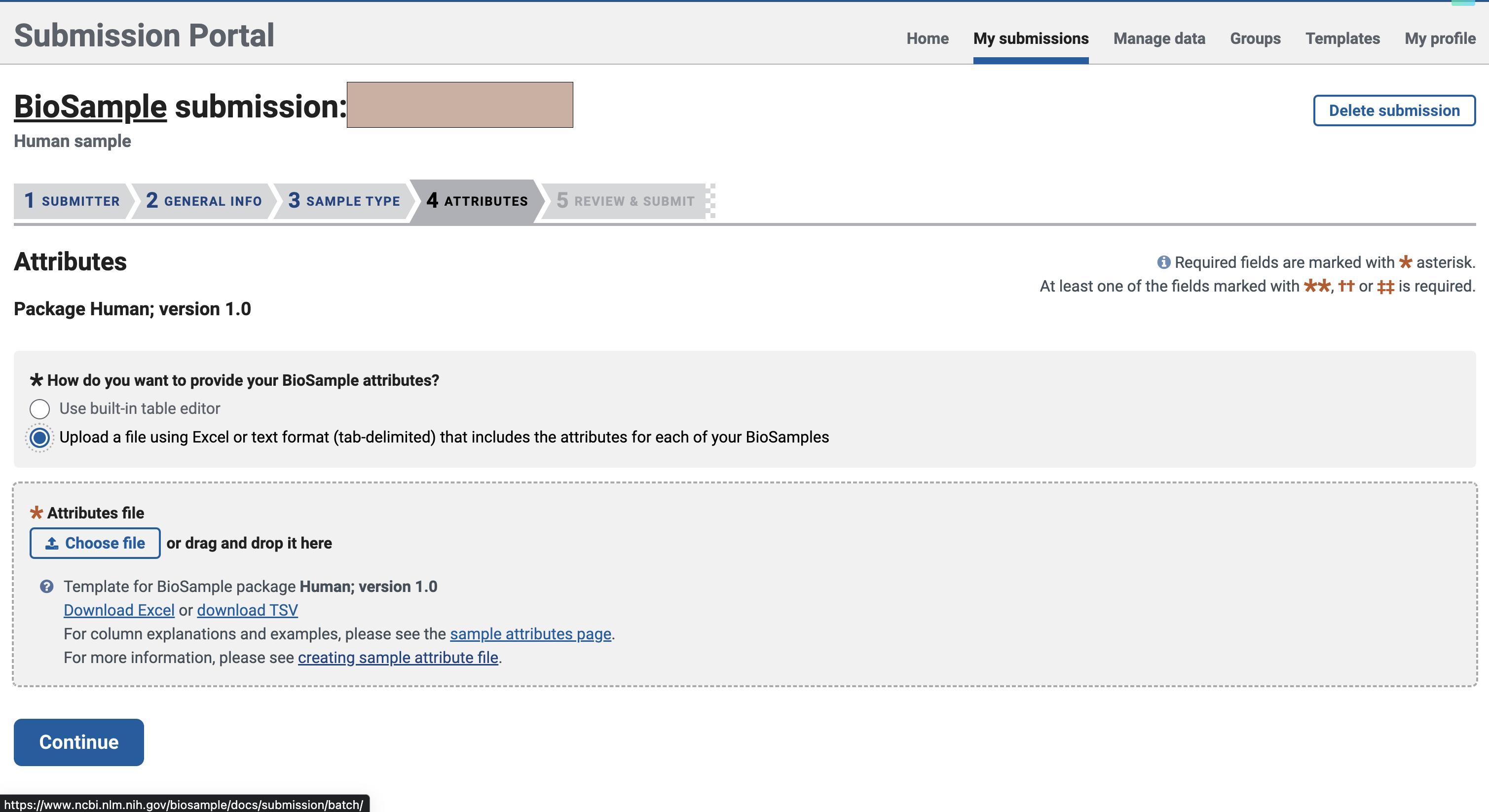
文件大概长这个样,绿色是必填项,黄色是可选项,你也可以添加自己的属性。不过需要注意的是,对于不同的sample_name,后面的属性不能完全一致,即使是同一种病,建议把病人来源标上,这样就能不重复了。
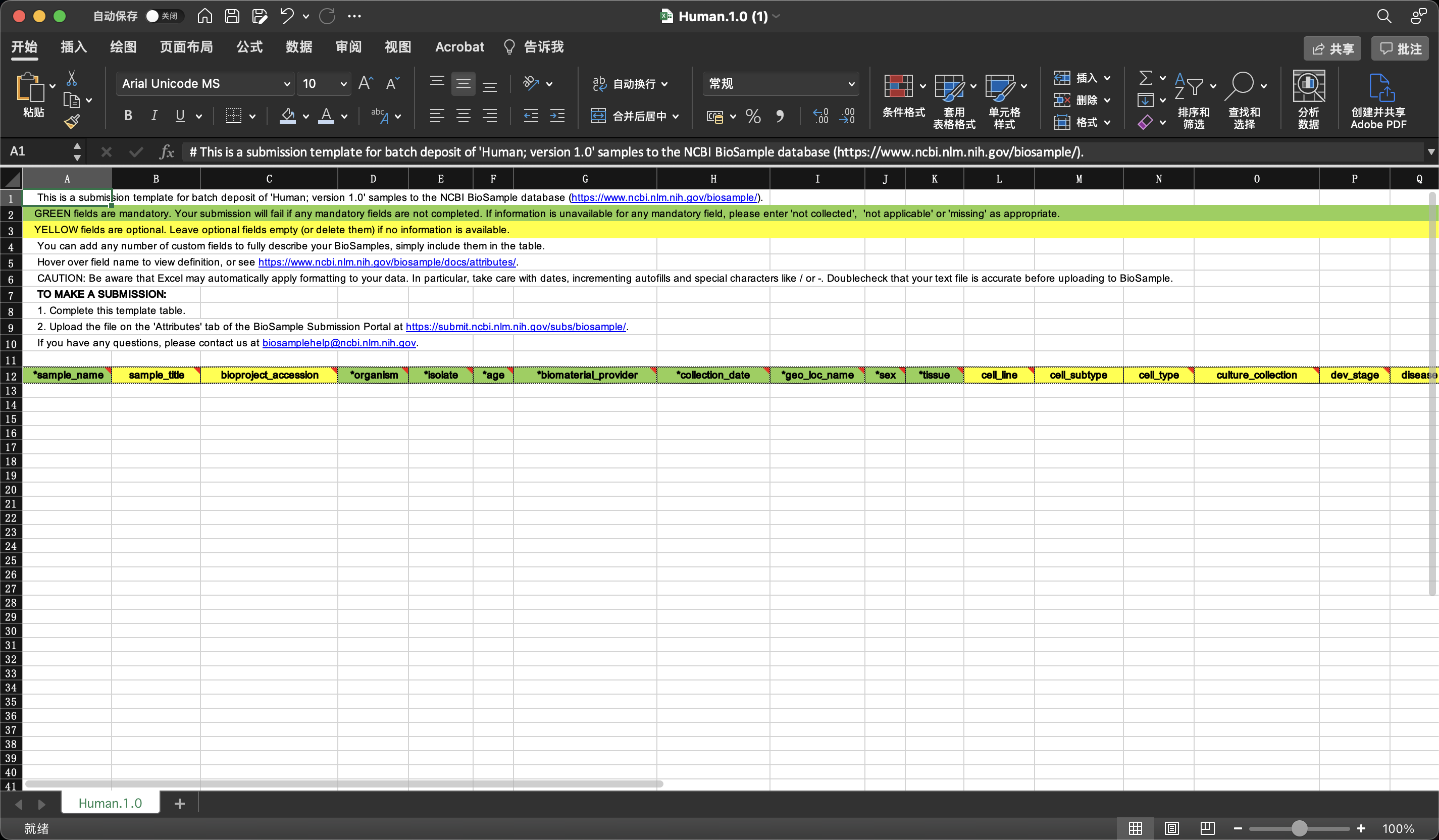
我们提交后出现这个页面,等待管理员审核即可。
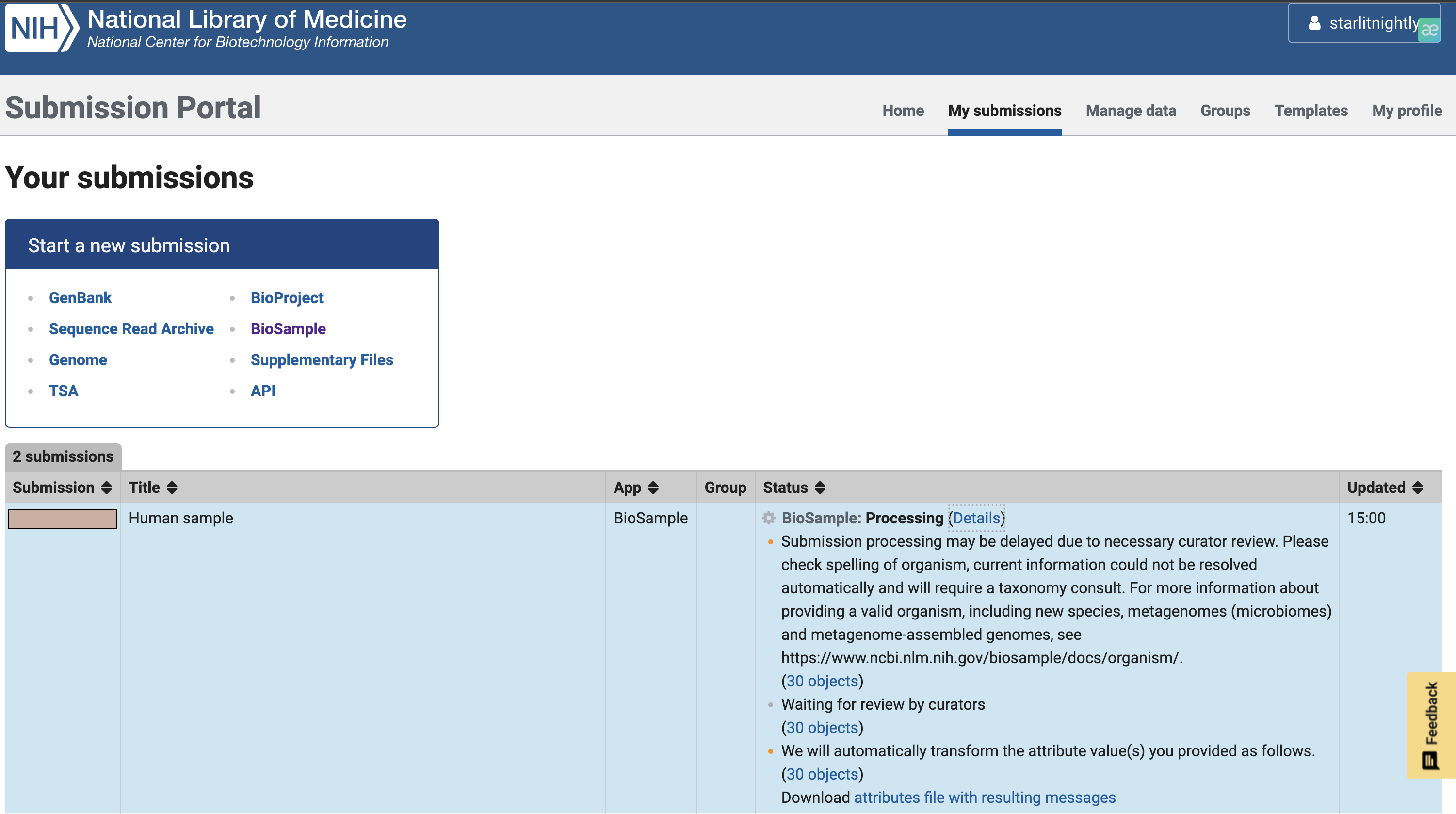
3. 上传fastq
3.1 biosample获取
经过三天的等待,我们的fastq的上传审核终于完成了
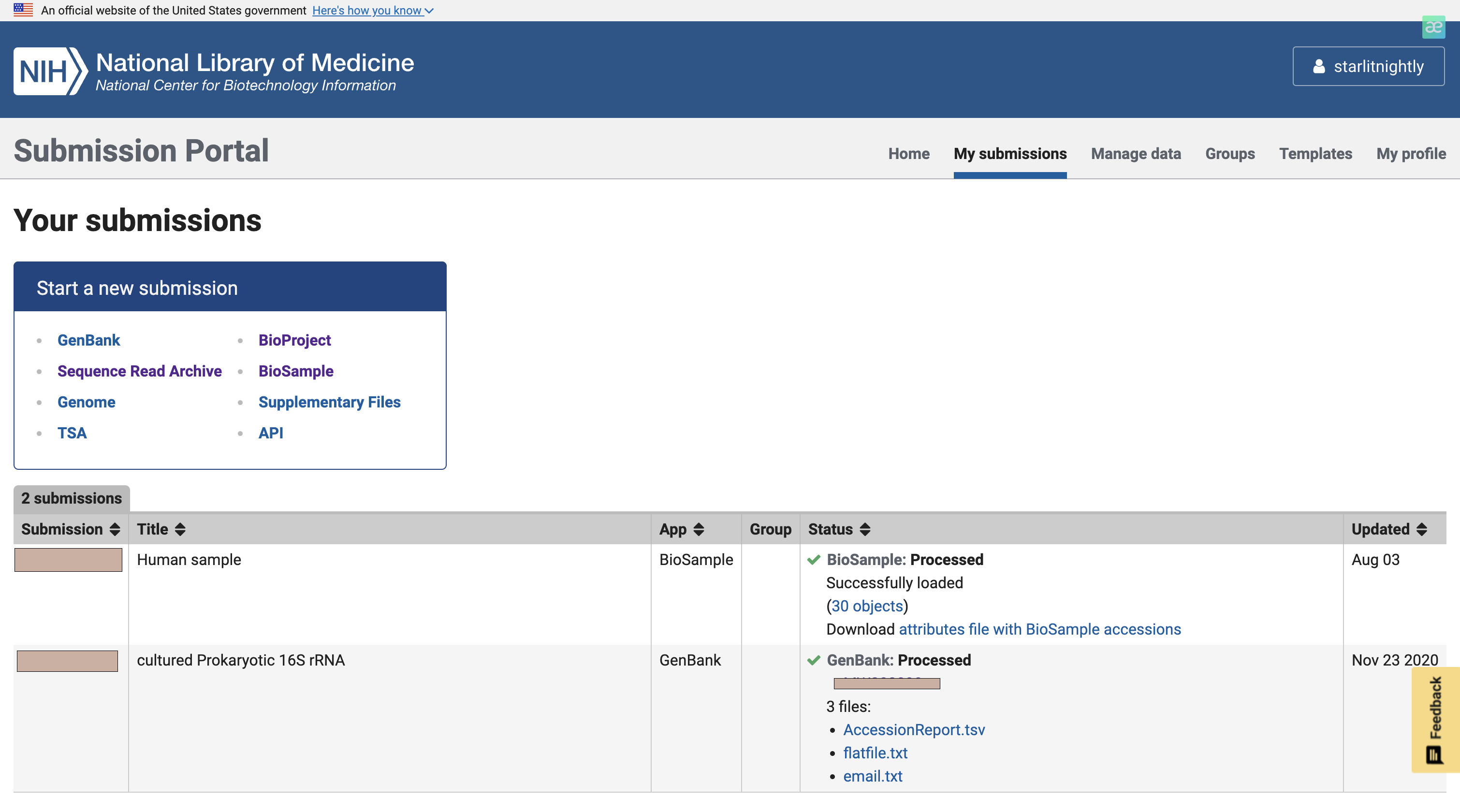
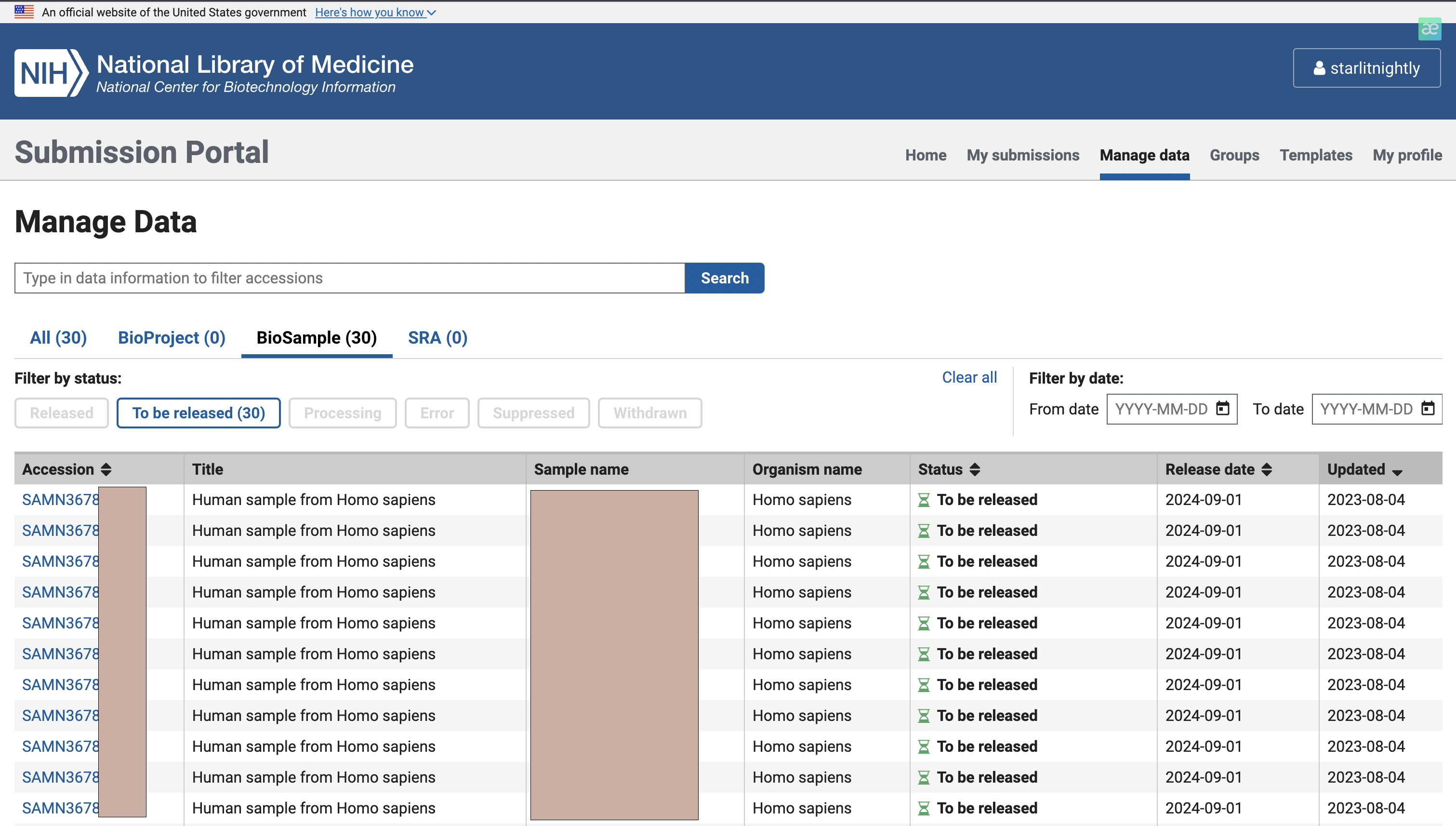
我们点击manage data发现多了30个biosample可以进行上传,这里的Accession就是我们的biosample的id,每个样本唯一。
3.2 上传SRA数据
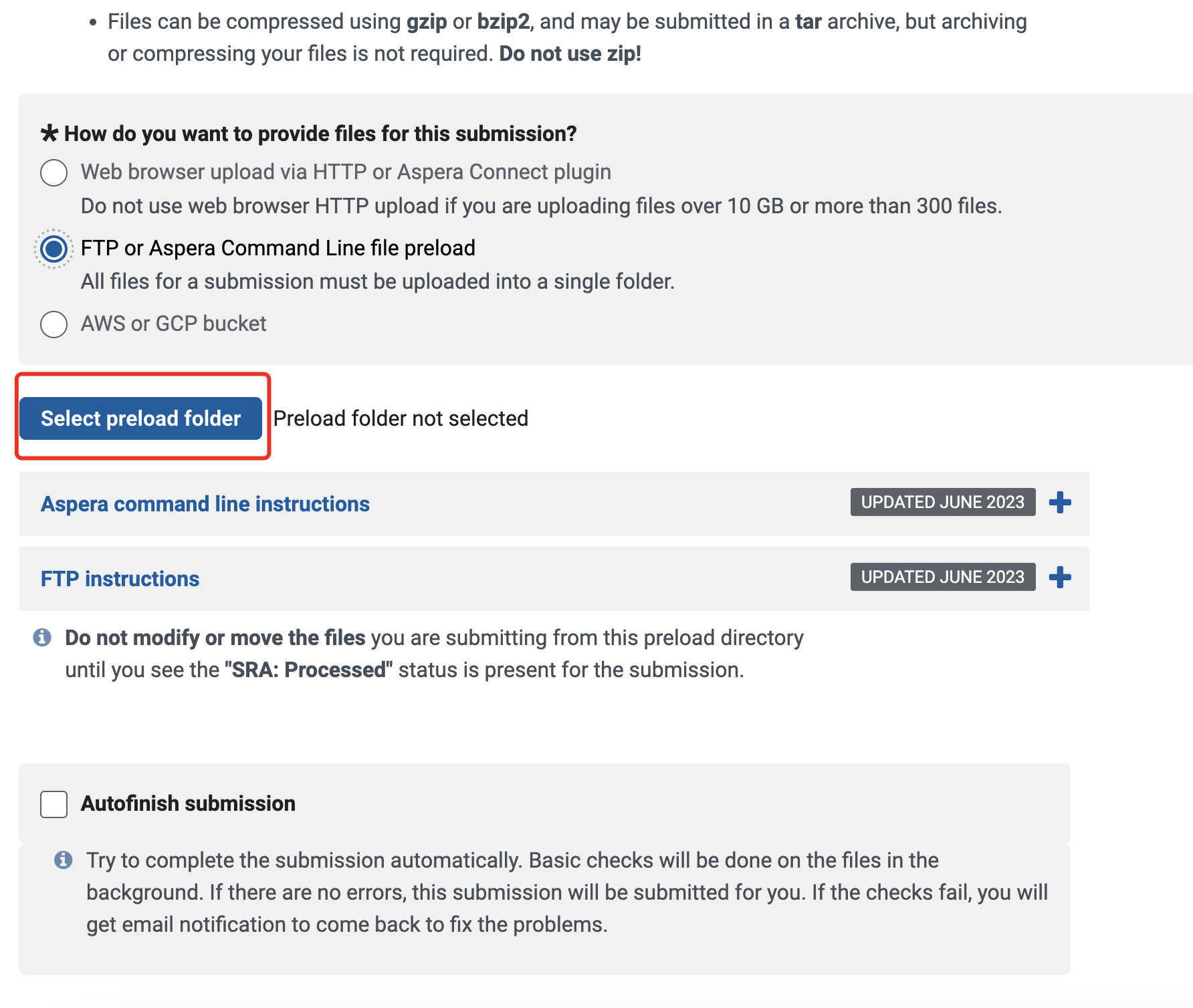
我们点击上一个页面的Sequence Read Archive,准备上传fastq
由于我的数据在服务器上,所以我们选择命令行上传
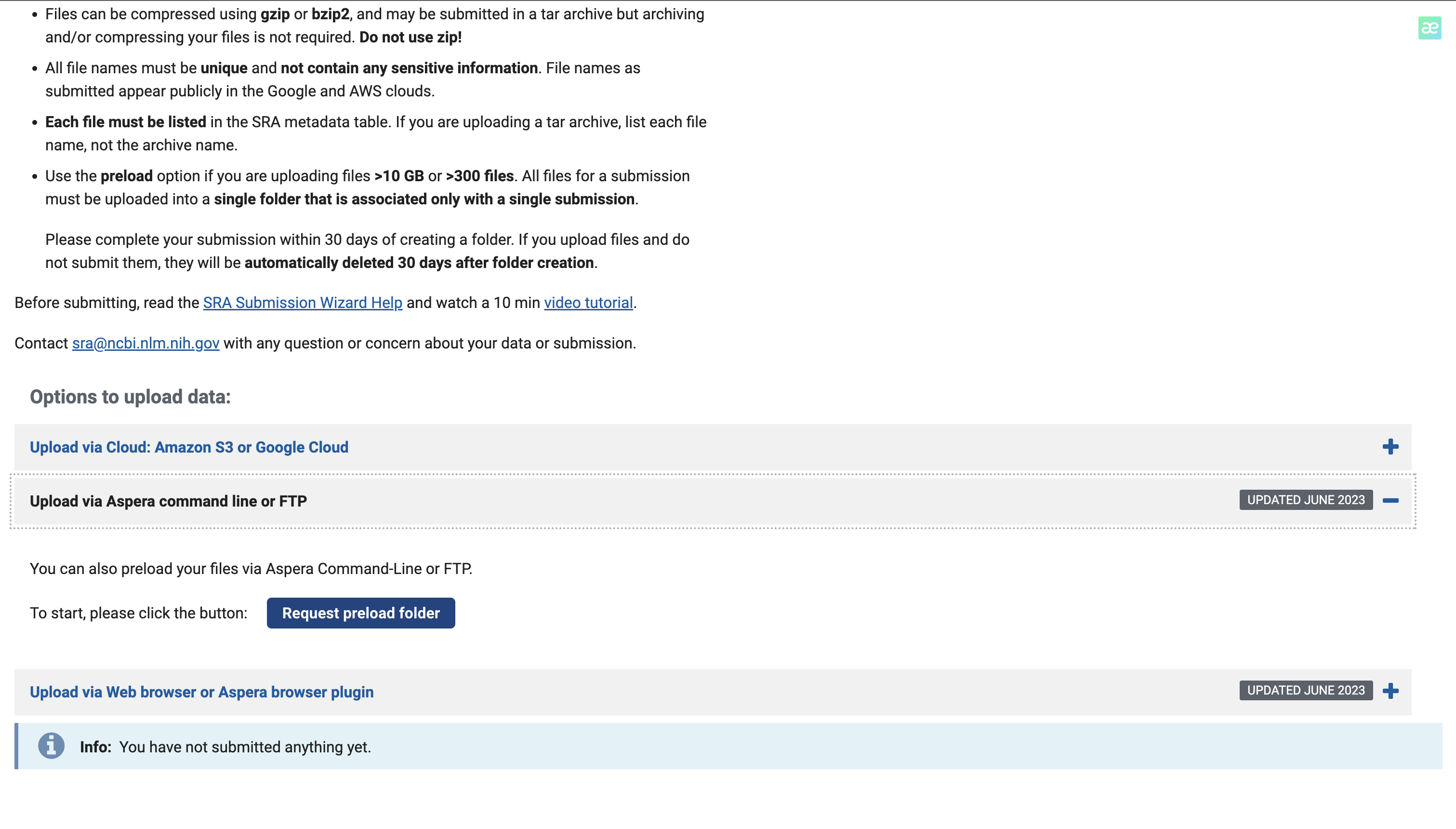
点击Request preload folder,ncbi提供了详细的上传教程
3.3 ascp环境安装
直接使用conda安装ascp,避免各种无意义的不兼容与报错。因为ascp的安装需要特定的glibc版本。
conda create -n ascp python=3.8
conda activate ascp
conda install -c hcc aspera-cli -y
ascp -h #检查安装
准备好ascp环境后,我们选择开始构建SRA项目
3.4 SRA上传内容填写
构建的过程比较简单,我们需要输入每一列所对应的信息即可,这里提供一个例子
| biosample_accession | library_ID | title | [library_strategy](#'Library and Platform terms'!A2) | [library_source](#'Library and Platform terms'!A27) | [library_selection](#'Library and Platform terms'!A36) | library_layout | [platform](#'Library and Platform terms'!A66) | instrument_model | design_description | filetype | filename | filename2 | filename3 | filename4 | filename5 | filename6 | filename7 | filename8 | assembly | fasta_file | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SAMN36786*** | Name | RNA-seq of Homo sapiens: Decidua | RNA-Seq | TRANSCRIPTOMIC SINGLE CELL | RANDOM | paired | ILLUMINA | Illumina NovaSeq 6000 | Single cell 10X | fastq | ***_1_1_R1.fq.gz | ***_1_1_R2.fq.gz | ***_2_1_R1.fq.gz | ***_2_1_R2.fq.gz | ***_3_1_R1.fq.gz | ***_3_1_R2.fq.gz | ***_4_1_R1.fq.gz | ***_4_1_R2.fq.gz |
其中,你下载的excel里面有很多是可以选的,所以我加粗的才是需要自己填写而不法选择的。
- biosample_accession: 生物样本访问号
- library_ID: 库ID,这个是独一无二的,需要自己想好用什么ID存在SRA数据库
- title: 数据标题,这个标题就任意,但是一般规则是RNA-seq of 物种:组织
- library_strategy: 库构建策略,这里一般是RNA-seq
- library_source: 数据来源库,这里一般是TRANSCRIPTOMIC SINGLE CELL
- library_selection: 库选择方式,这里一般是RANDOM
- library_layout: 库布局,我这里是双端测序,所以写paired
- platform: 测序平台,我从测序公司给的文件里找到了Illumina
- instrument_model: 仪器模型,我从测序公司给的文件里找到了Illumina NovaSeq 6000
- design_description: 设计描述,我们是单细胞测序所以是Single cell 10X
- filetype: 文件类型,我们上传的文件类型,这里一般都是fastq
- filename: 文件名(第1个文件)一般一个单细胞文库最大是8个文件,双端测序的话最小是2个文件,大部分都是两个文件,取决于测序公司给你的结果。
- filename2: 文件名(第2个文件)
- filename3: 文件名(第3个文件)
- filename4: 文件名(第4个文件)
- filename5: 文件名(第5个文件)
- filename6: 文件名(第6个文件)
- filename7: 文件名(第7个文件)
- filename8: 文件名(第8个文件)
3.5 上传本地fastq
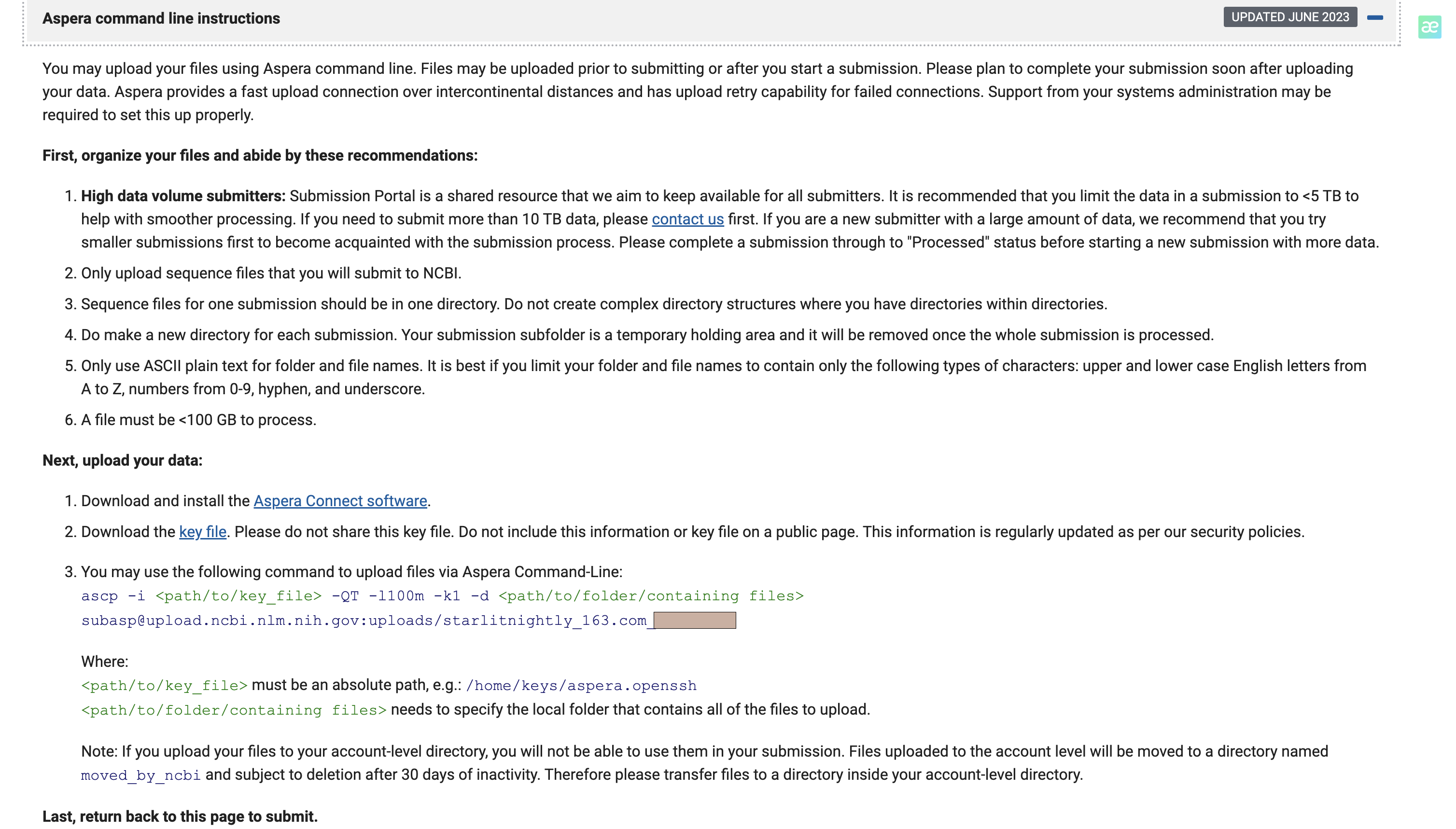
填好这个表后,我们点击continue,进入文件上传页面,跨洲际上传,这里只介绍ascp,以下命令是从官网复制的,切记不可照抄
ascp -i /mnt/home/zehuazeng/ncbi/aspera.openssh -QT -l100m -k1 -d /mnt/home/zehuazeng/media/207D/origData subasp@upload.ncbi.nlm.nih.gov:uploads/starlitnightly_163.com_***
这里需要修改的只有-i和-d两个参数,注意是绝对路径
- -i: openssh文件路径,这个是在上传页面有一个超链接可以下载
- -d: 需要上传的文件夹,里面只需要包含你要传的fq文件即可,文件名与前面填的filename1-8一致。
由于网络波动,所以我写了一个.sh文件,检测到上传失败可以自动重传,我们命名该文件为upload.sh
#!/bin/bash
while true; do
ascp -i /mnt/home/zehuazeng/ncbi/aspera.openssh -QT -l100m -k1 -d /mnt/home/zehuazeng/media/207O/origData8 subasp@upload.ncbi.nlm.nih.gov:uploads/starlitnightly_163.com_UFgOBYes
if [ $? -eq 0 ]; then
echo "Command completed successfully."
break
else
echo "Command failed. Retrying in 5 seconds..."
sleep 5
fi
done
然后在终端输入./upload.sh即可运行,注意文件需要先修改权限chmod 777 ./upload.sh。
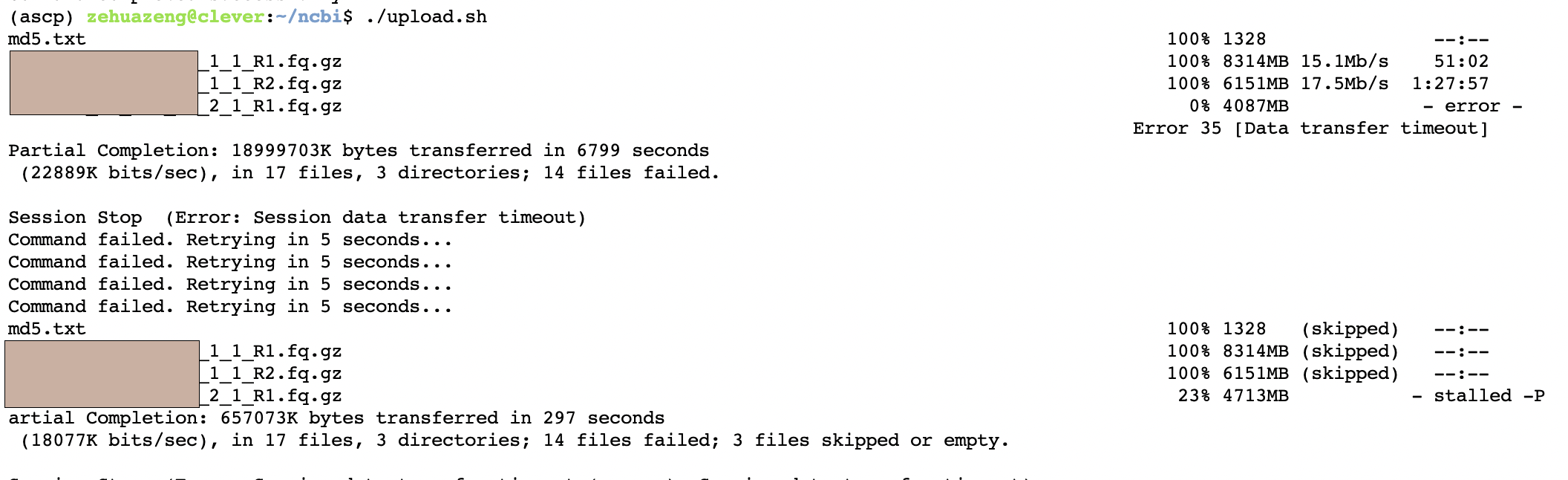
我们发现自动上传便开始了,并且会自动检测上传成功与失败。我们等全部文件上传好后,回到刚才的SUB页面
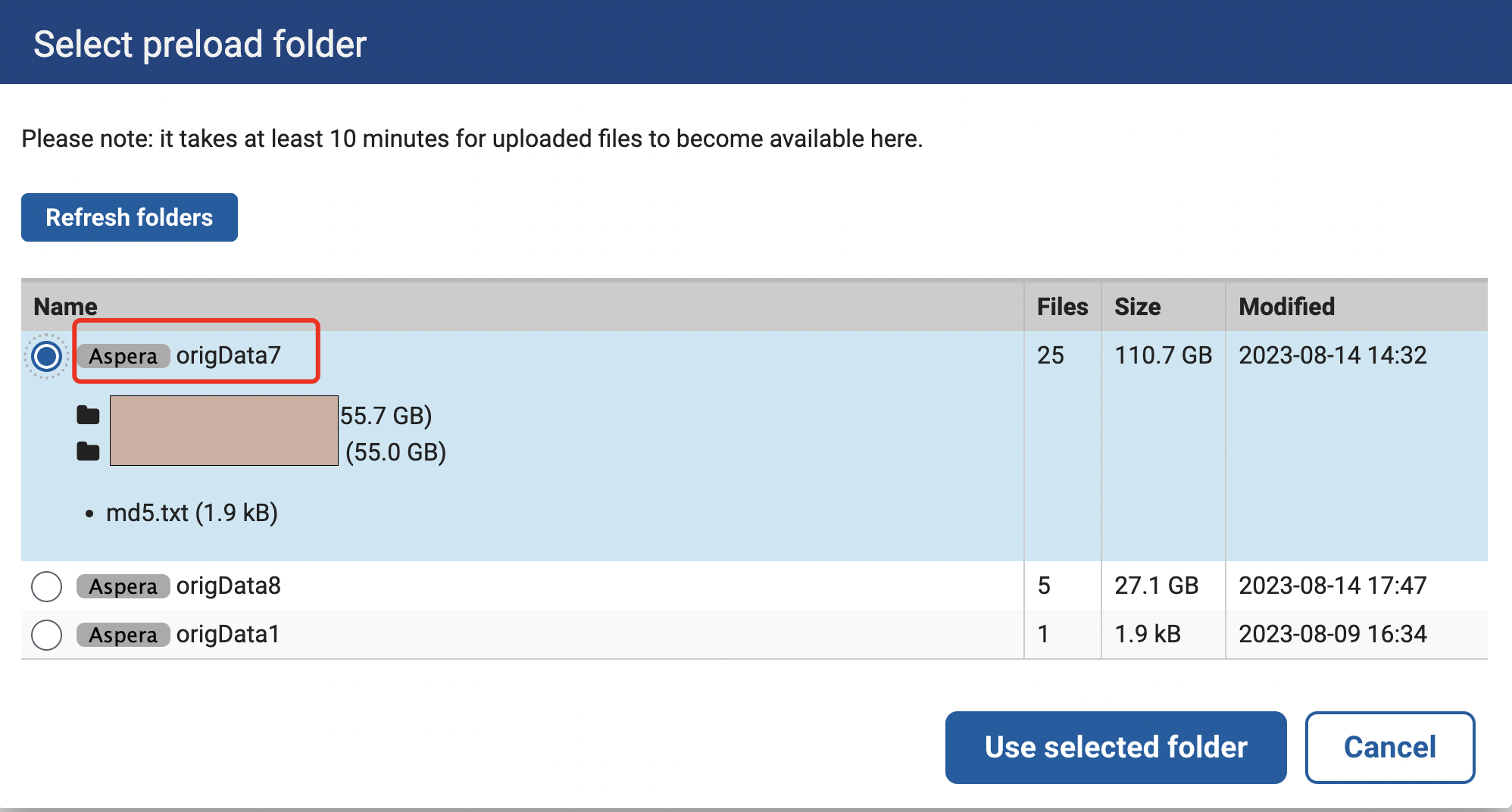
我们选择Select preload folder,这里origData7是我们已经传好的文件夹,而origData8是正在上传的文件夹
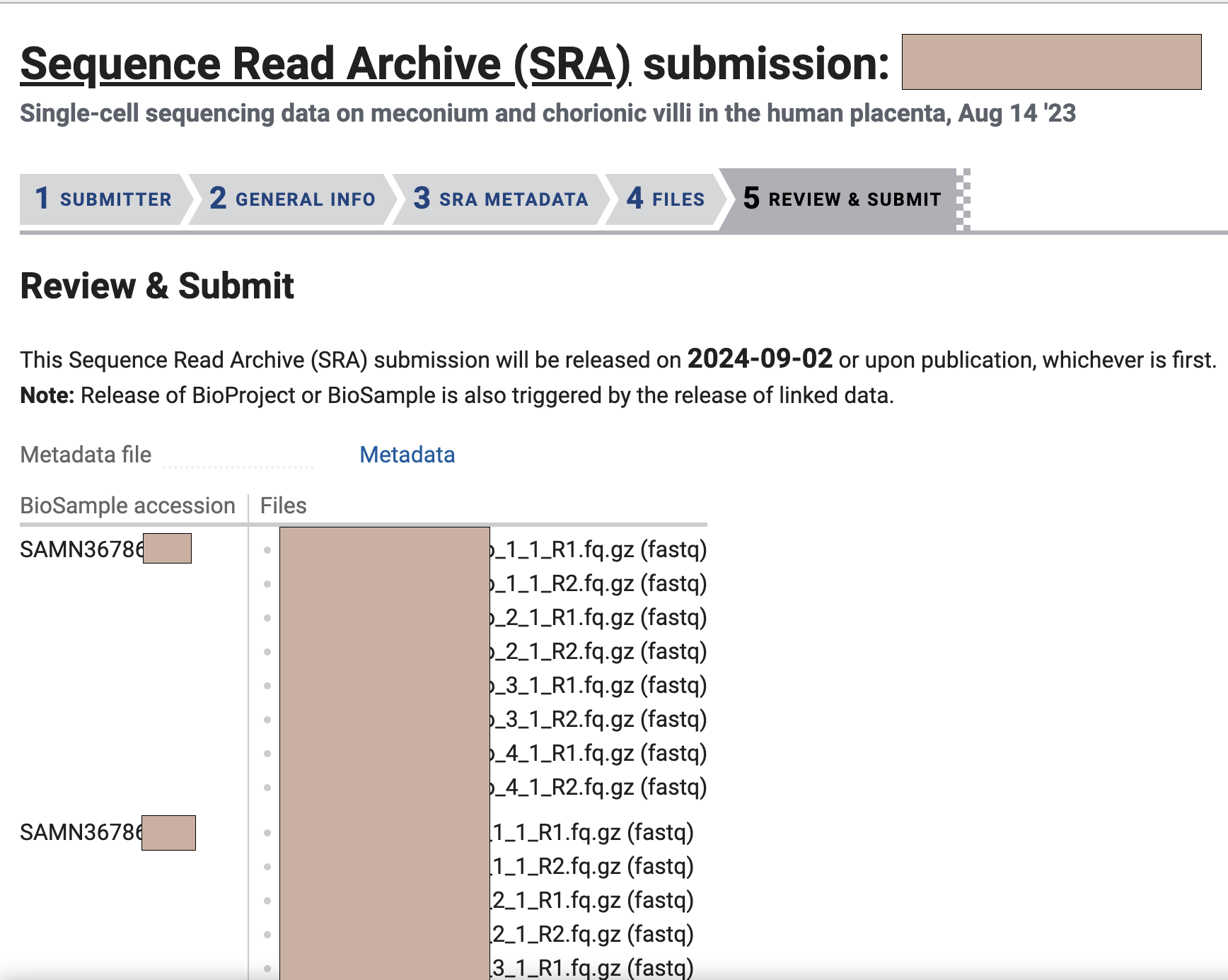
选择完了我们点击Continue,就会跳到最后一个页面,一般来说文件会自动帮你选出来,根据你前面填写的filename1-8
我们点击Submit就好了,会跳转到最开始的页面,提示我们正在处理中。
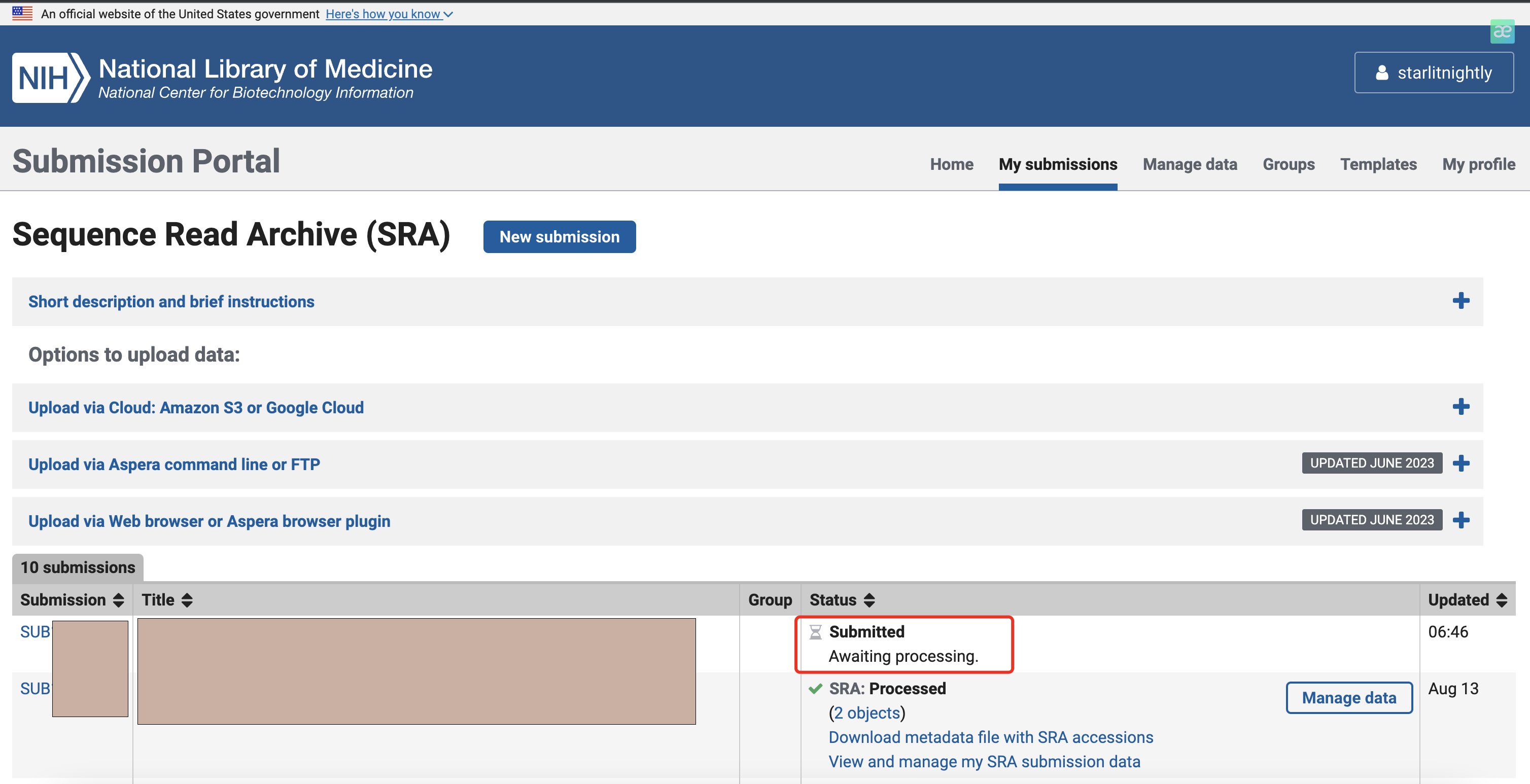
3.6 其他
我在上传的时候遇到了一次失败,报错提示是
2023-08-06T22:03:02 sra_subprocess error: Finished: /panfs/traces01.be-md.ncbi.nlm.nih.gov/trace_software/pipeline/sra_prod/transform_tools/sharq_load.py --config /panfs/traces01.be-md.ncbi.nlm.nih.gov/trace_software/pipeline/sra_prod/config.sra.public /panfs/traces01.be-md.ncbi.nlm.nih.gov/trace_software/pipeline/sra_prod/transform_tools/sharq --platform=ILLUMINA --log-level=info --output=/export/home/SSD/production_sra_public/sge1240.212826.trace.run_load.sh/SRR25541***.run_load/SRR25541***.output ***_3_1_R2.fq.gz ***_4_1_R2.fq.gz ***_2_1_R2.fq.gz ***_2_1_R1.fq.gz ***_1_1_R1.fq.gz ***_1_1_R2.fq.gz ***_3_1_R1.fq.gz ***_4_1_R1.fq.gz; pid=218120, rc=243
这种错误不是我能解决的,于是我写了封邮件发送到sra@ncbi.nlm.nih.gov,然后过了两天再看邮箱,工作人员后台已经帮我弄好了。
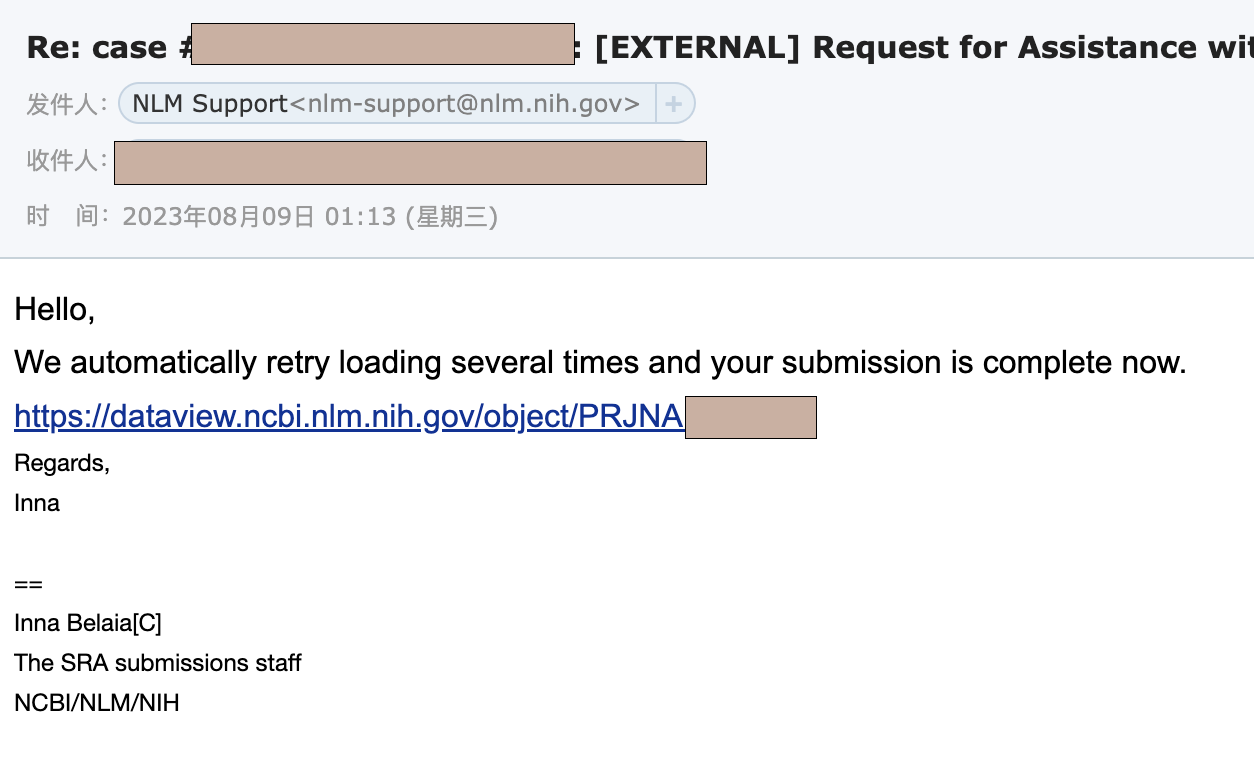
4. GEO上传
我们将原始数据成功上传到SRA数据库后,我们还需要上传处理后的数据到GEO数据库上。GEO数据库与SRA的上传有一些类似,但也有所区别。可能是由于处理后的文件通常不会太大,所以上传可以用ftp。
4.1 新建GEO提交
我们点击New Submission新建一个提交
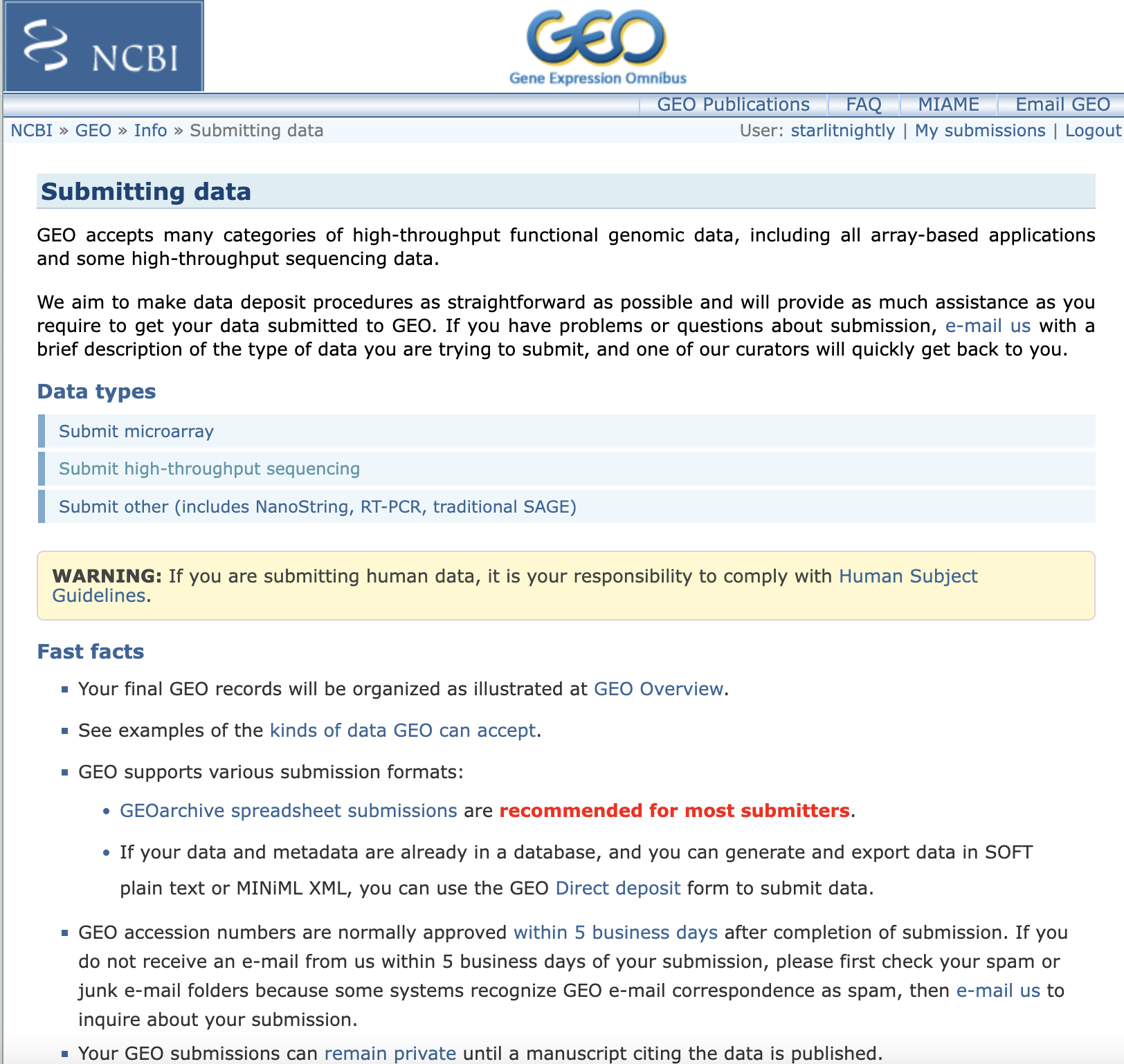
我们选择high-throughout sequencing来完成scRNA-seq数据的上传,点开后发现,我们需要先下载一个meta文件进行信息的填写。
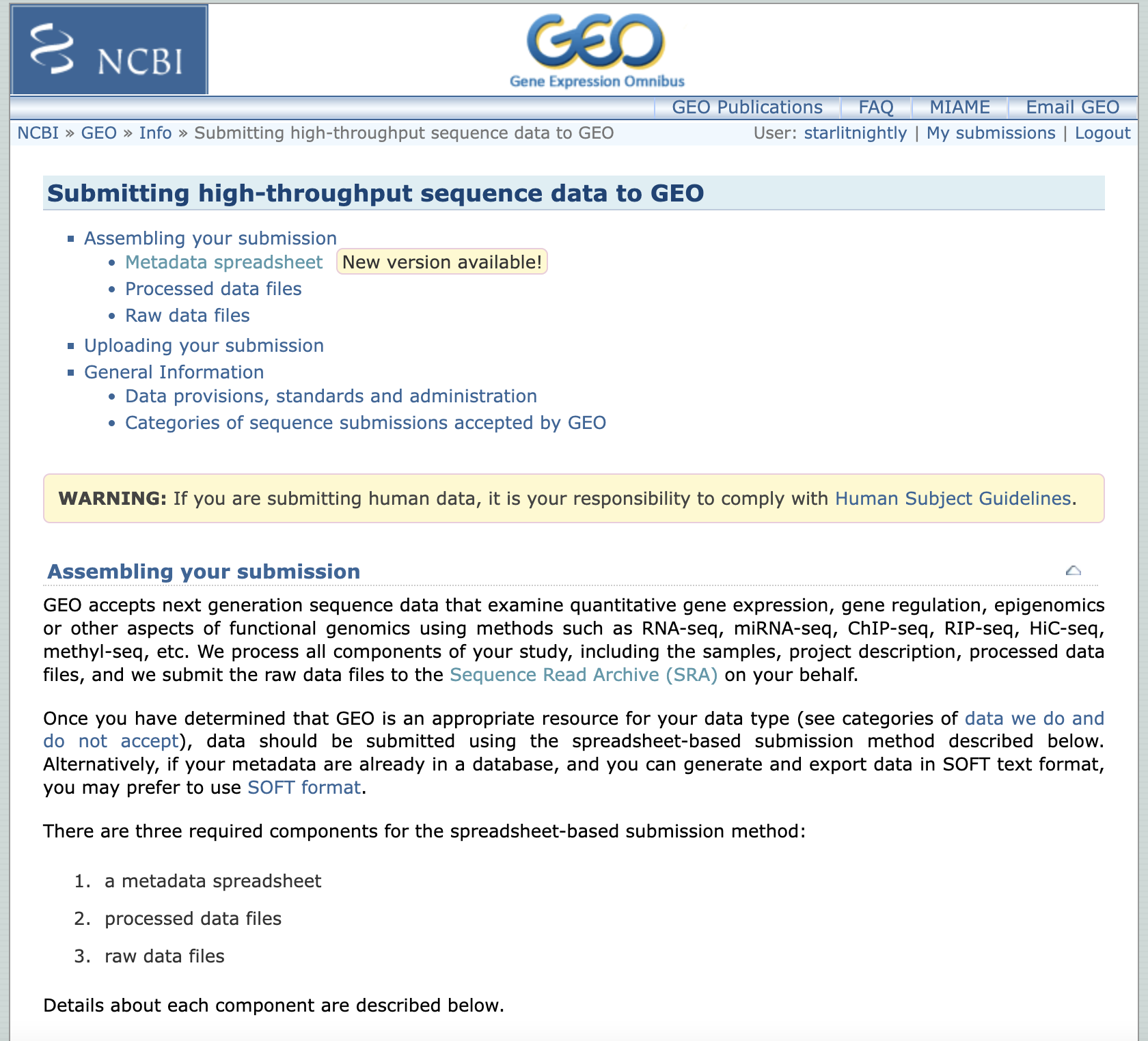
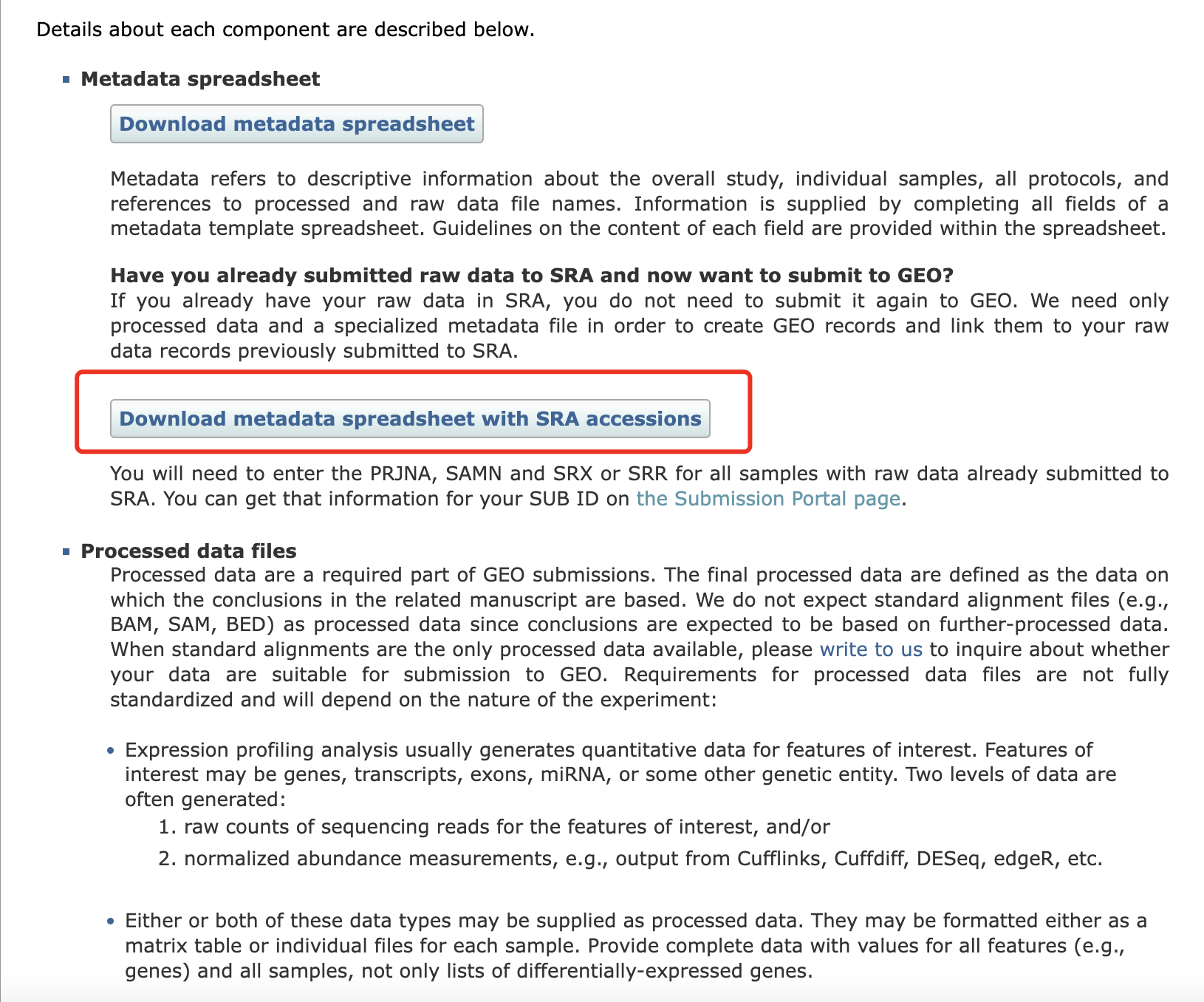
我们选择第二个,因为我们已经把测序文件上传到了SRA数据库,这样可以避免重复上传原始数据。
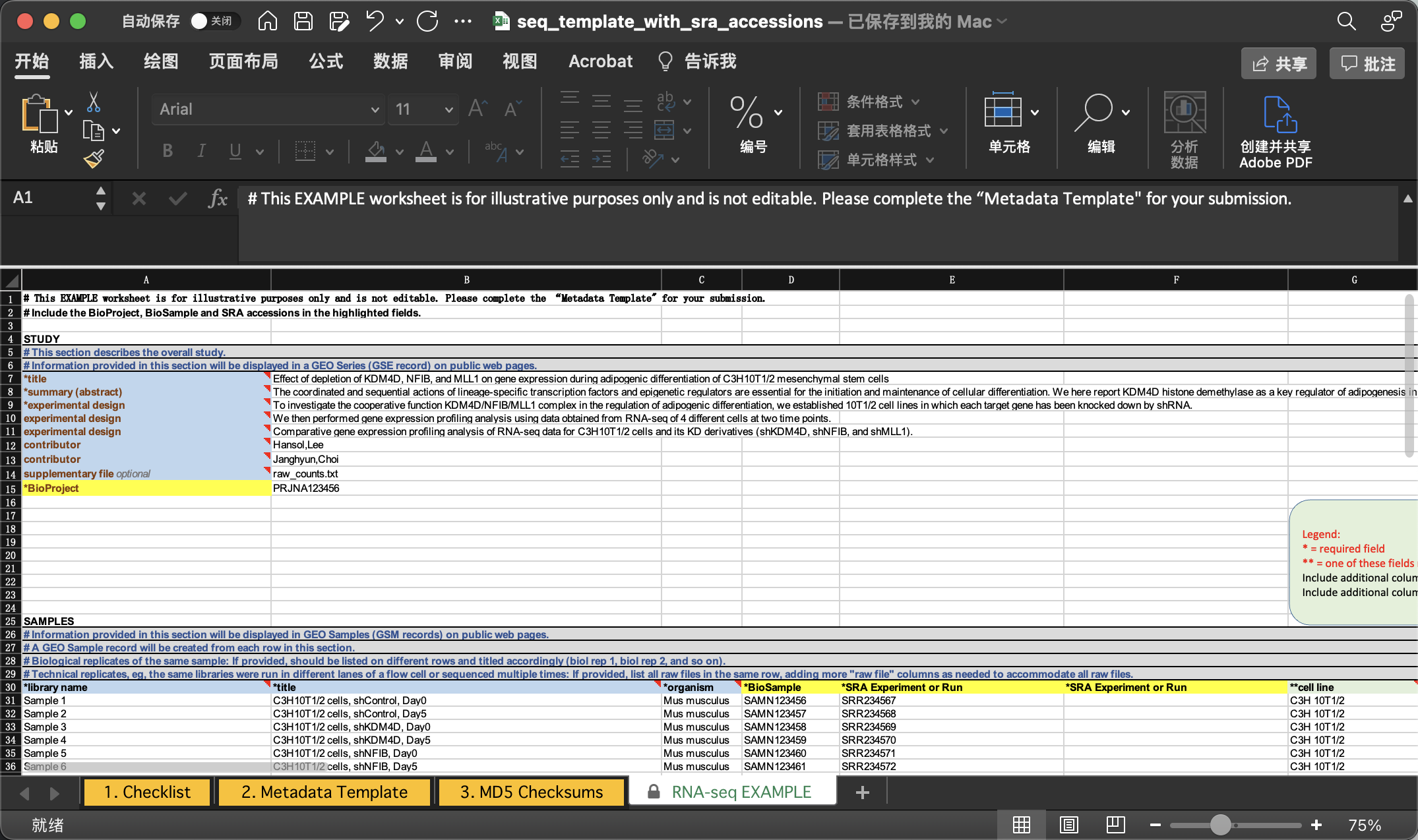
我们照着EXAMPLE的格式填写即可。
4.2 上传处理文件与meta
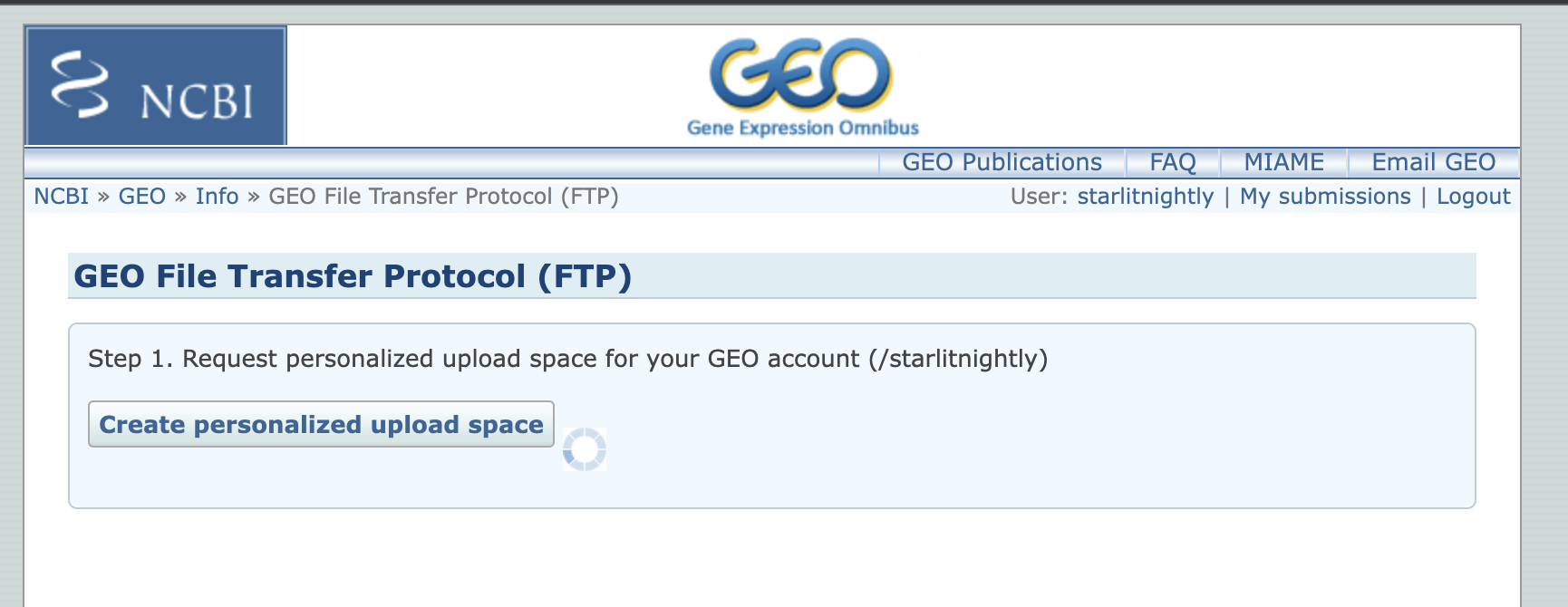
然后我们点击Create personalized upload space创建自己的ftp空间
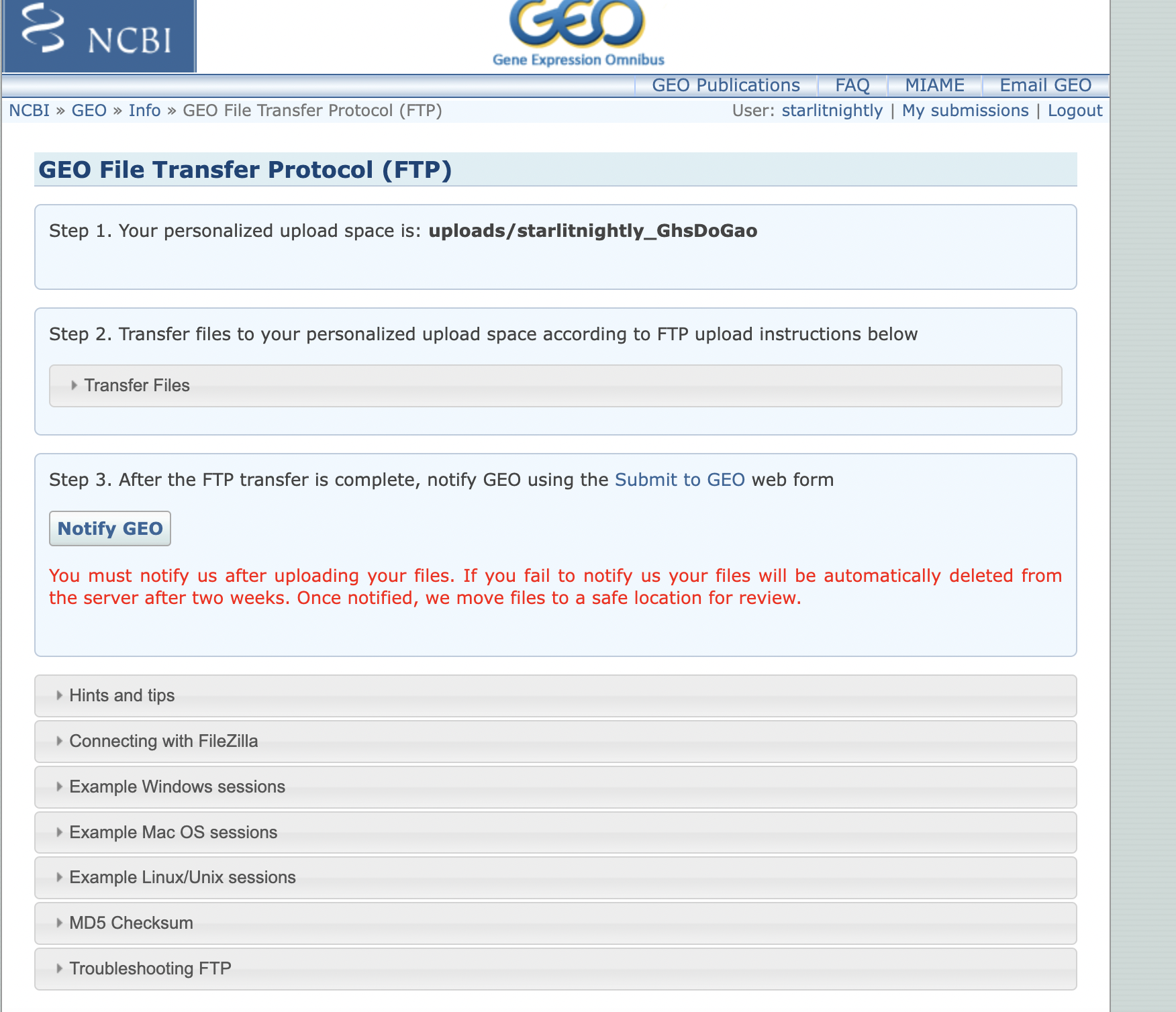
等待一会儿便会加载出Step 2,我们点击左边的箭头展开,会发现里面提供了GEO数据库的服务器信息。
- Host address:
- username
- password
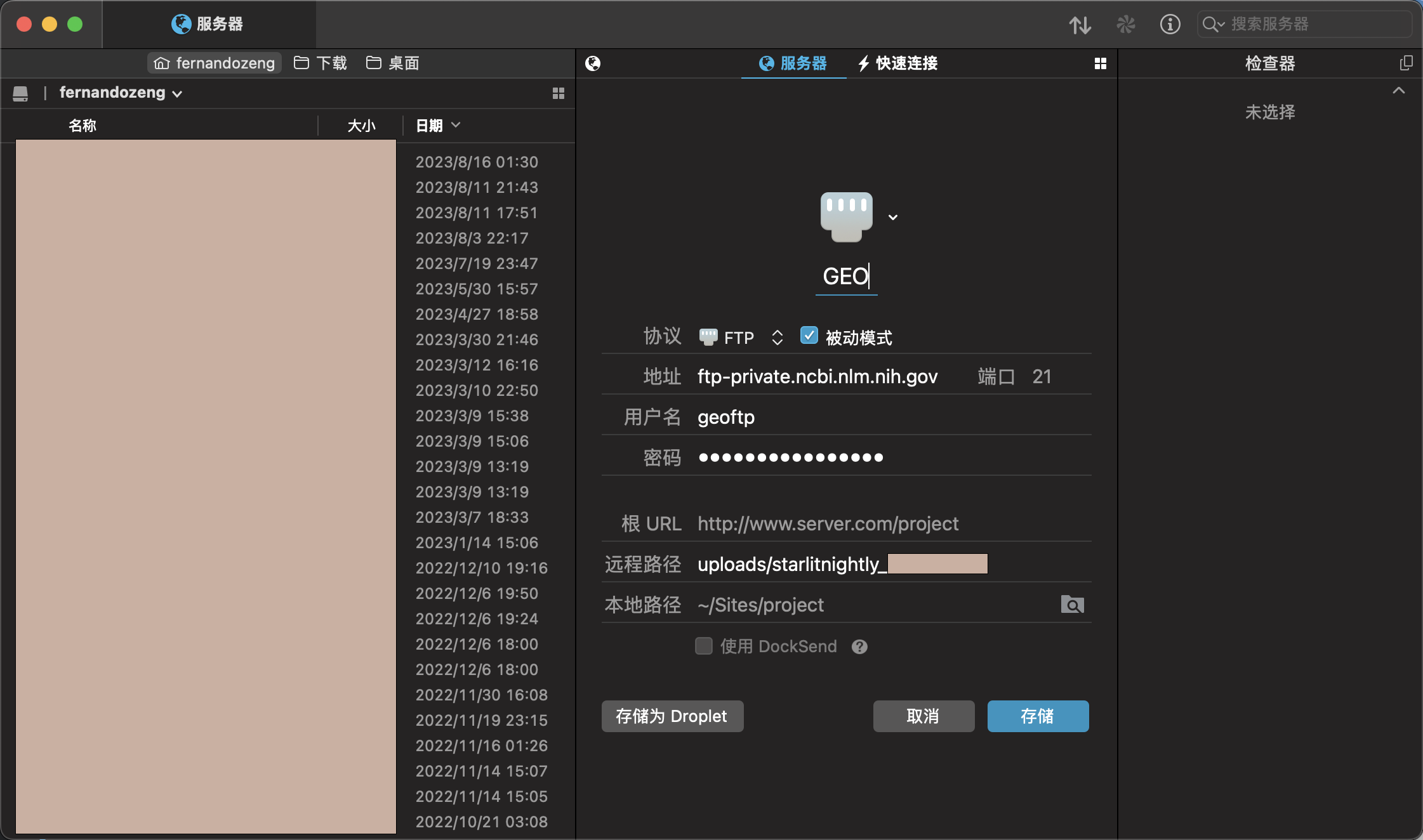
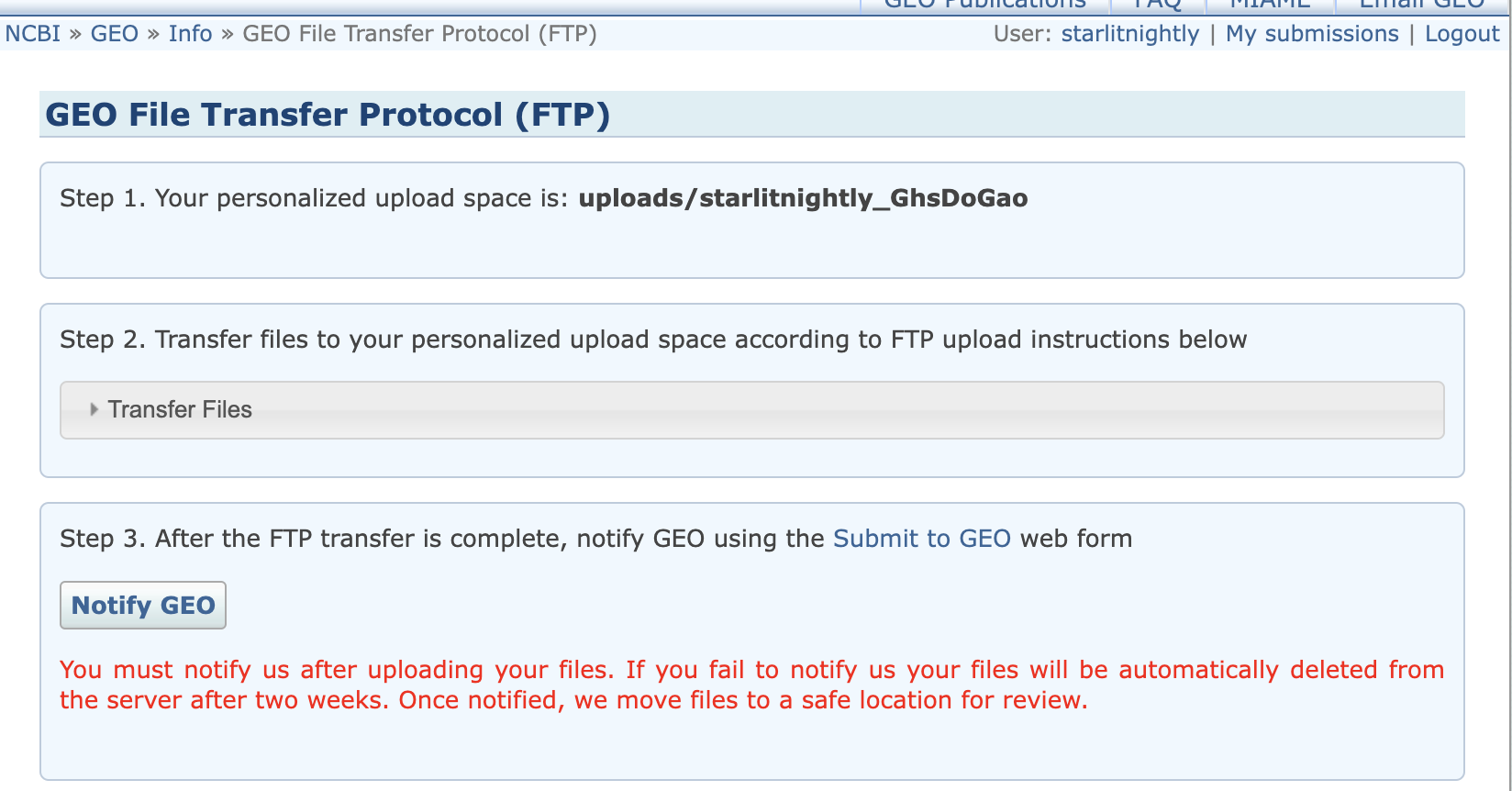
我们根据这三个信息可以连接到远程的GEO服务器上,但需要注意的是,我们连接的远程目录不能是默认的,而是uploads/starlitnightly_***这个在图中我用红色区域圈了起来

我是mac系统,所以我用transmit来做ftp文件传送,我们将刚才的Hostname填到地址,username填到用户名,password填到密码,同时设定远程路径为刚刚提到的uploads/starlitnightly_***
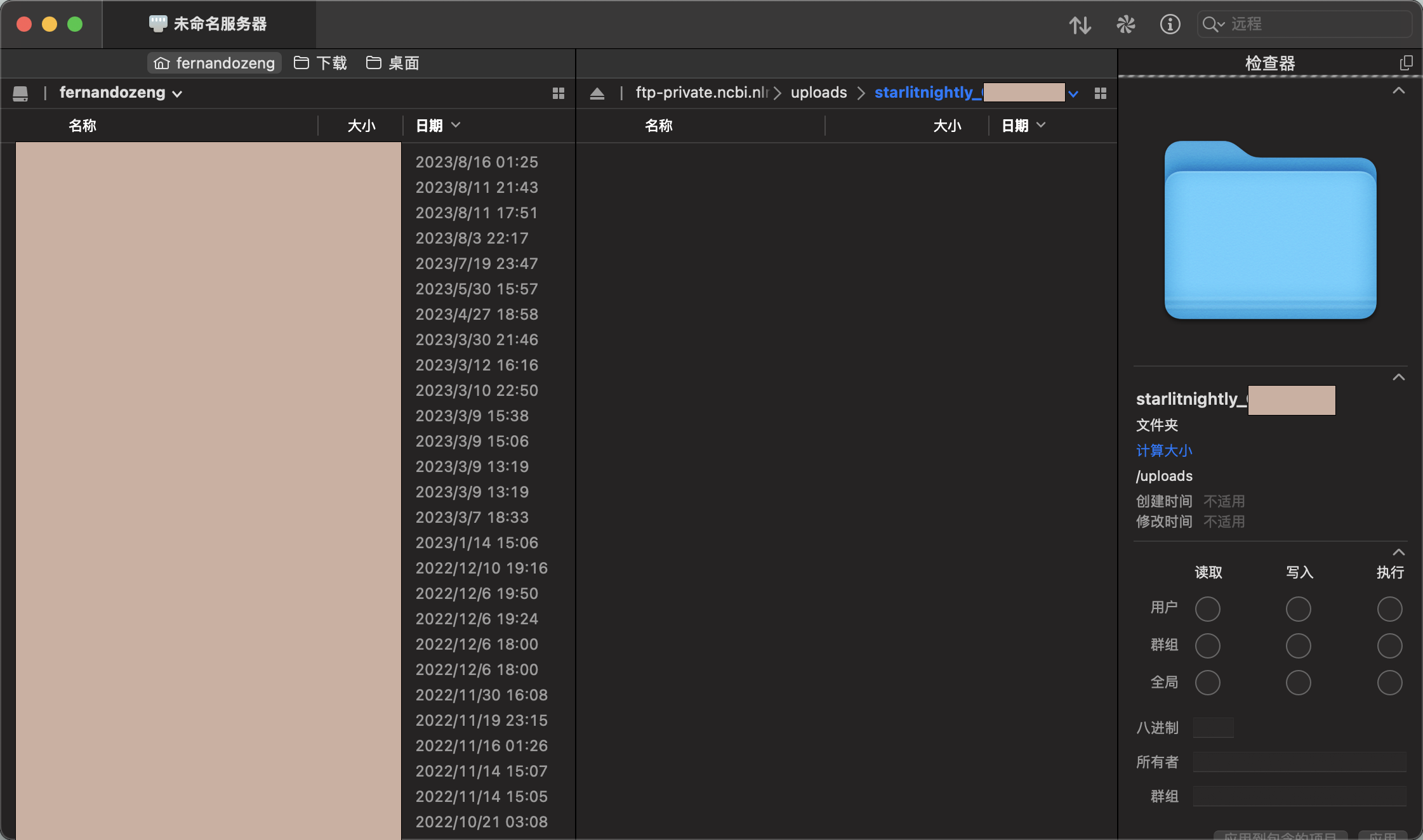
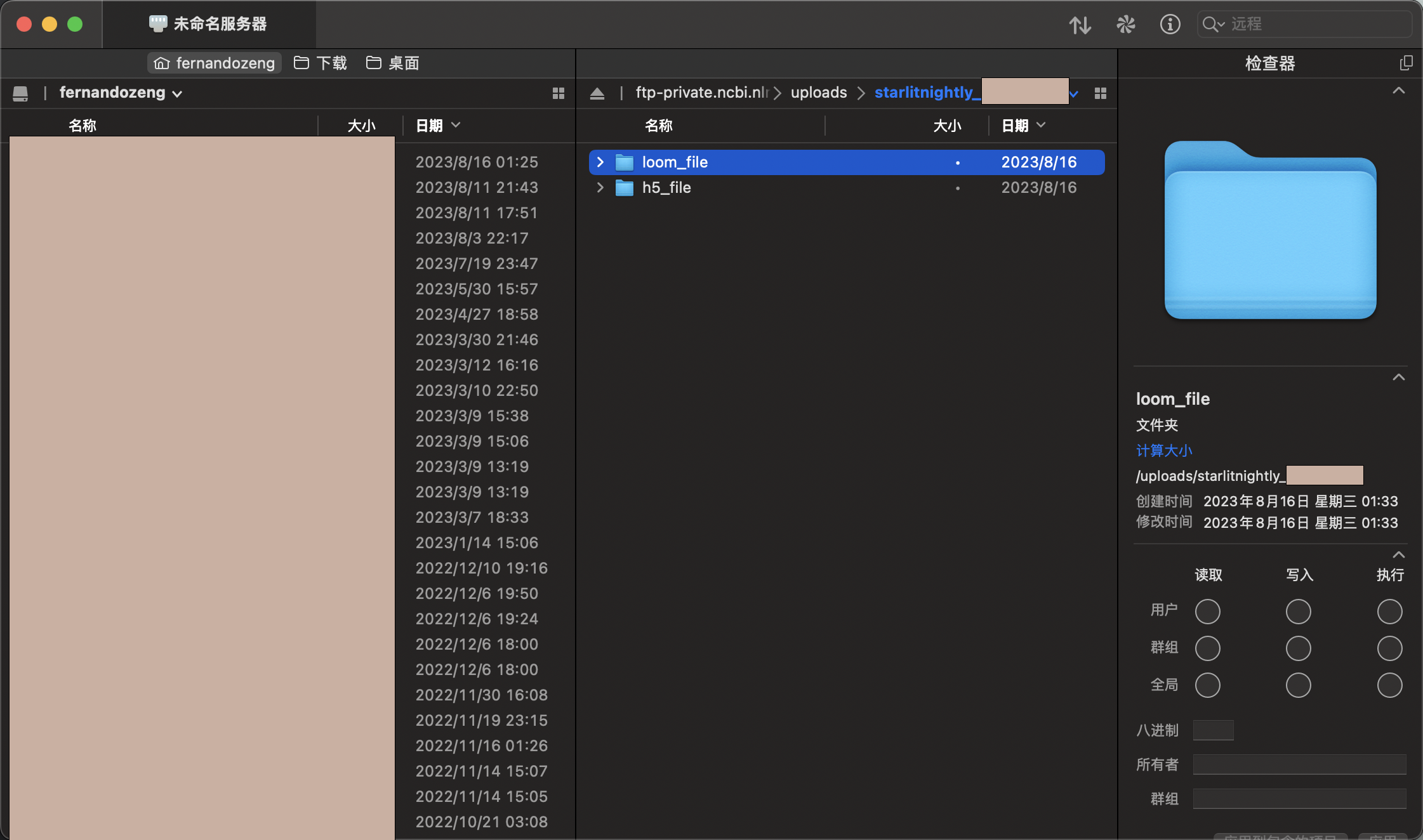
连接后我们会发现里面是空的,我这里需要传过滤后的h5文件以及velocyto生成的剪接/未剪接矩阵,所以我新建了两个文件夹,一个叫h5_file,一个叫loom_file,同时我在meta里面已经填写好了每一个样本对应的文件名
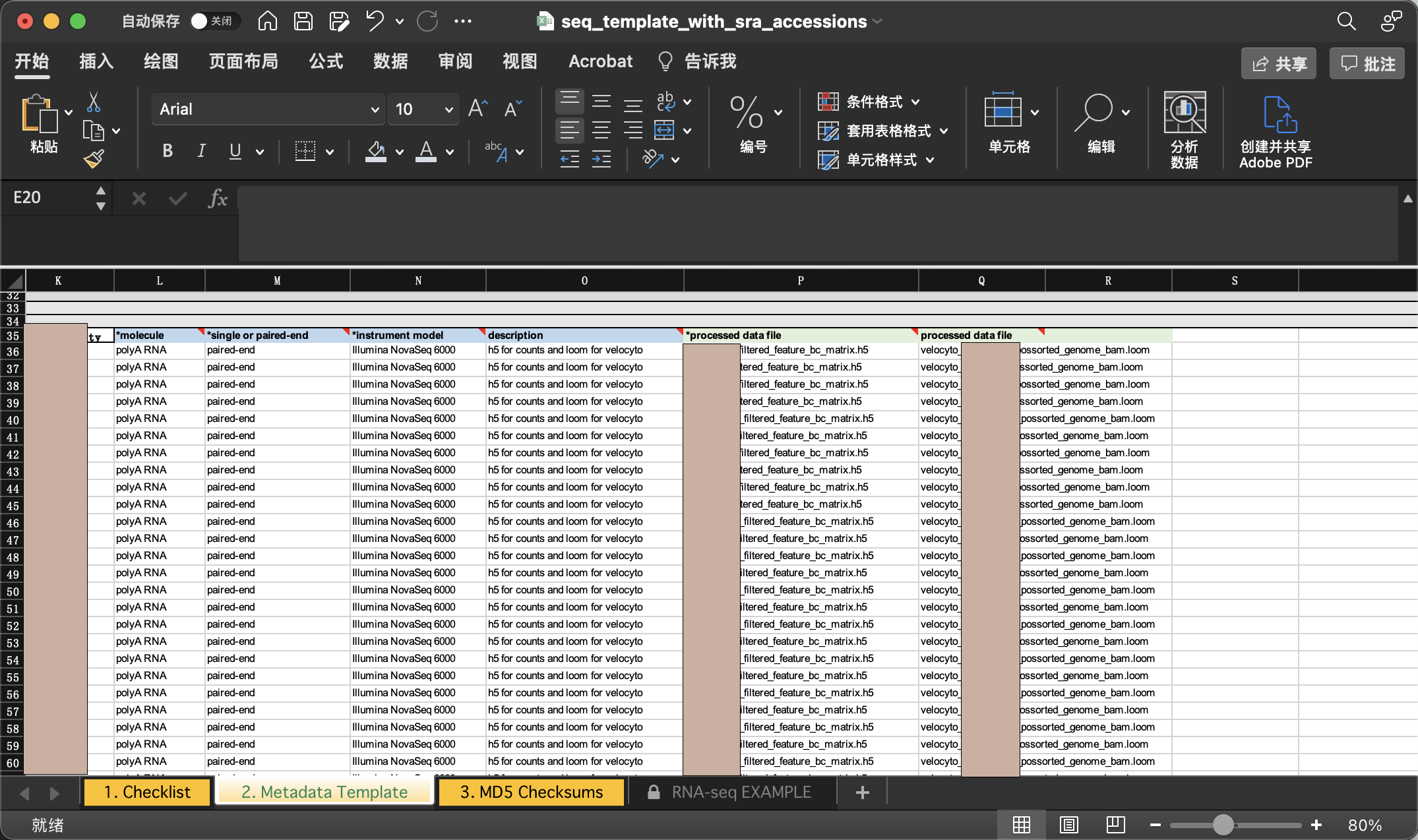
我们直接拖动文件进入对应文件夹即可上传。同时需要注意的是,meta文件也需要一并上传
4.3 提交GEO申请
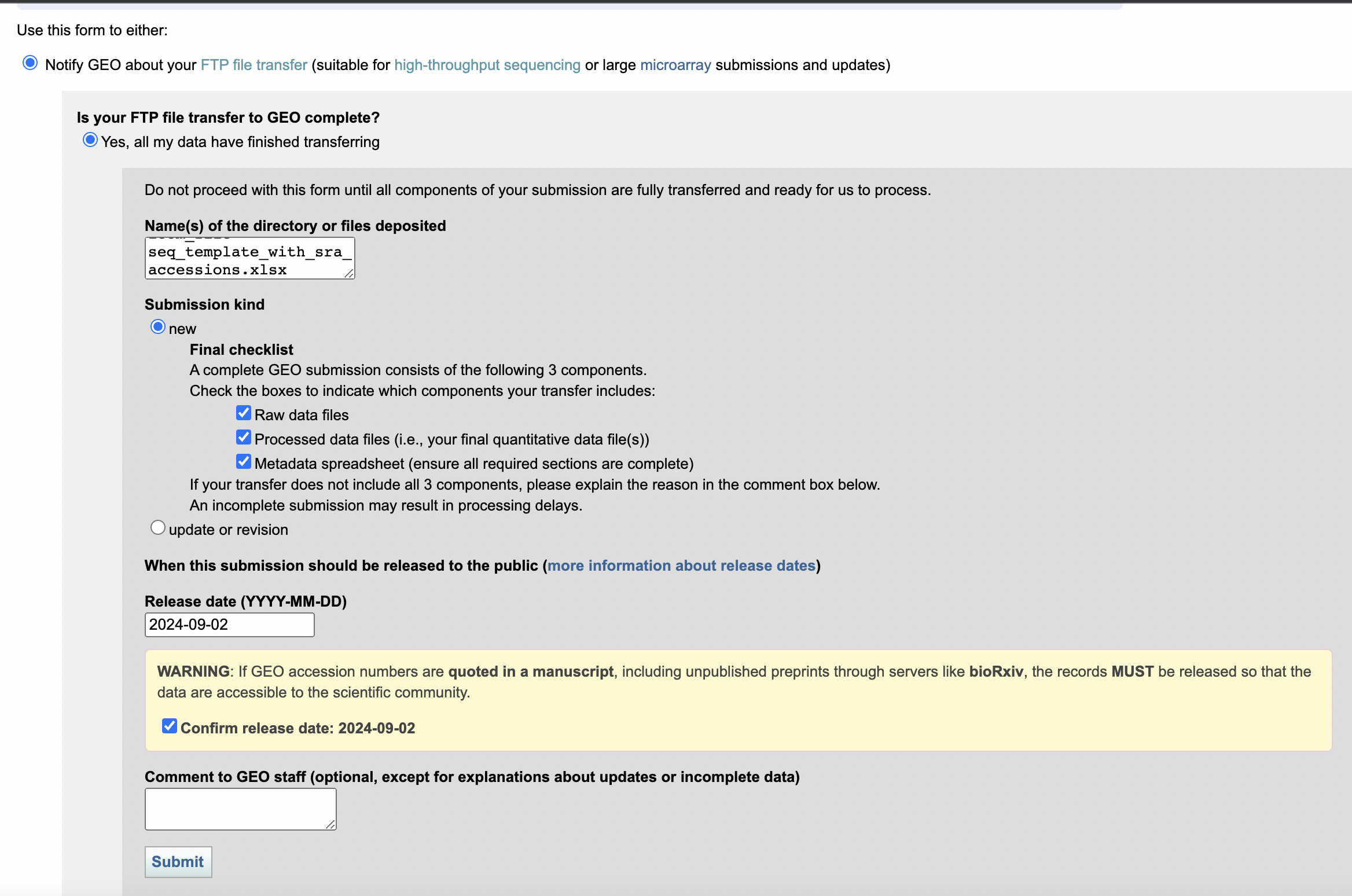
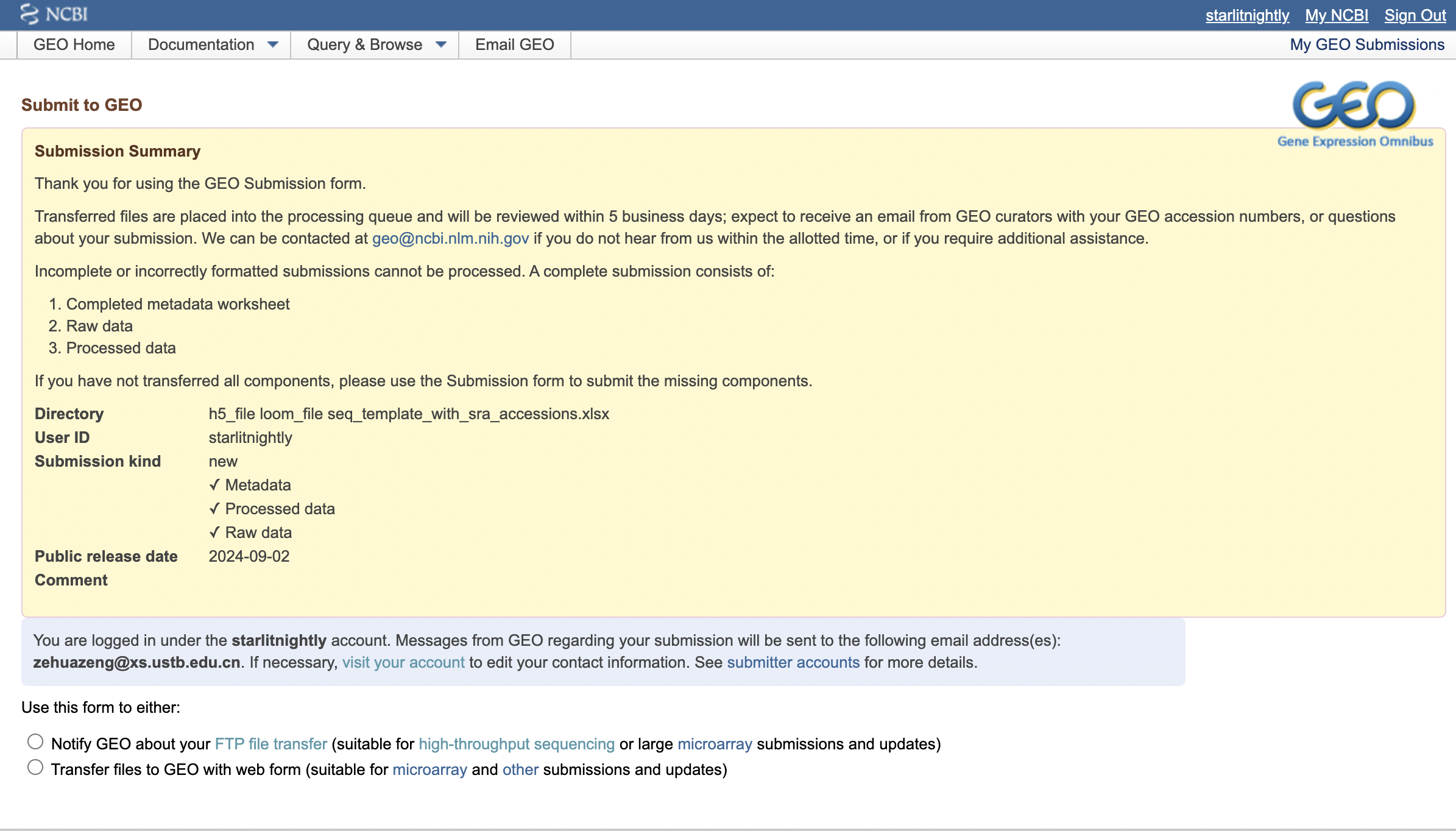
我们在传好文件后,回到GEO申请界面,点击Notify GEO,我们填写好目录和文件描述,点击submit即可。
4.4 确认GEO
大概一天左右(工作日),你就会收到一封来自GEO工作人员的审核邮件,如果有问题会在邮件里说明,我这里没有问题就直接回道GEO里查看我的提交,发现确实多了一个记录。
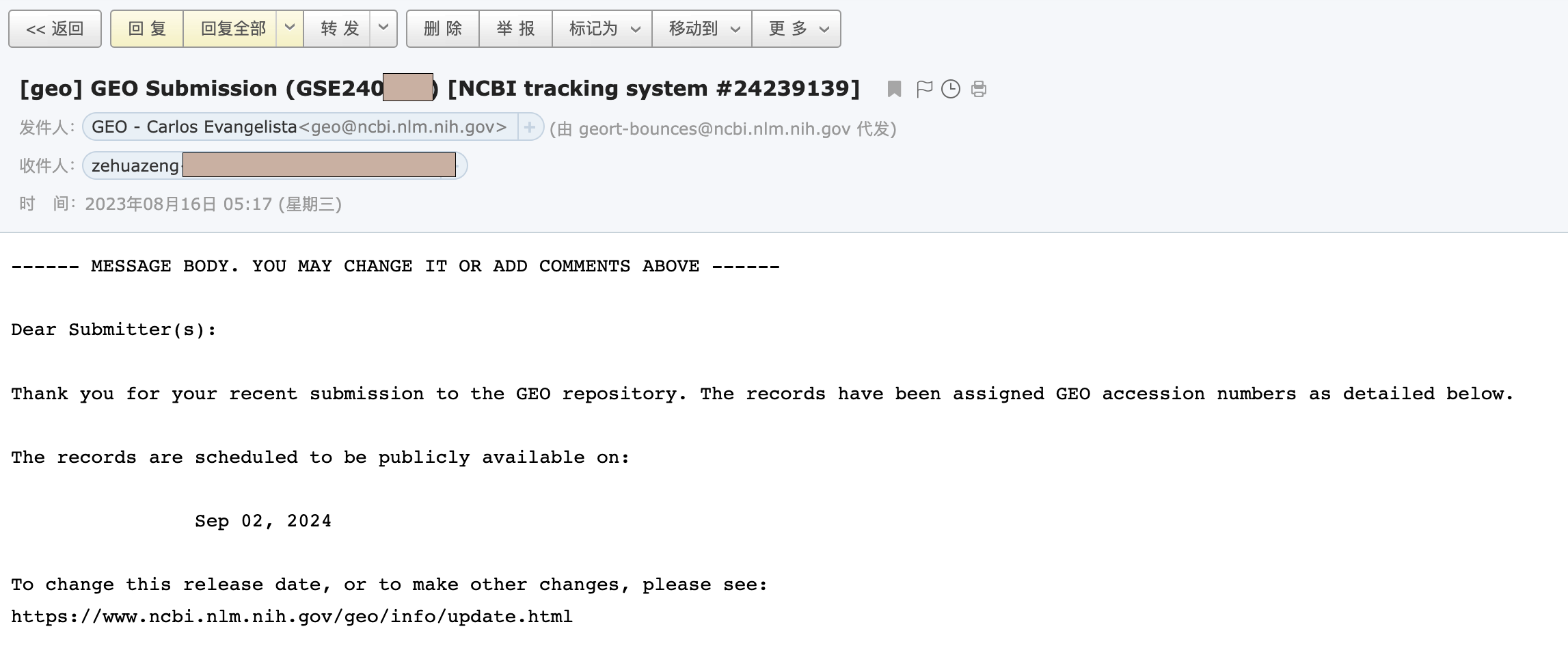
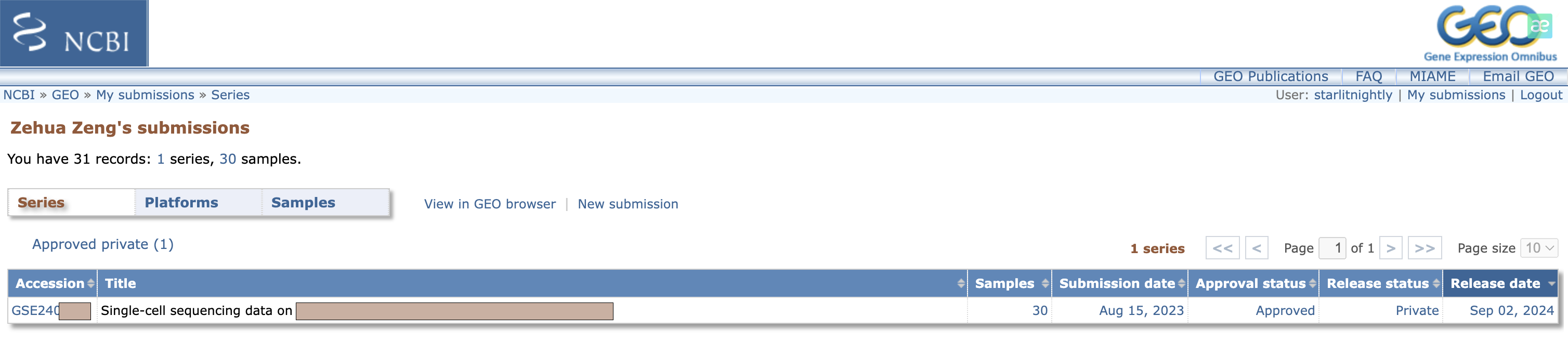
我们点进去后,可以点击Update更新一些相关信息。

单细胞测序最好的教程(十四)测序原始数据公开至NCBI数据库的更多相关文章
- 无废话ExtJs 入门教程十四[文本编辑器:Editor]
无废话ExtJs 入门教程十四[文本编辑器:Editor] extjs技术交流,欢迎加群(201926085) ExtJs自带的编辑器没有图片上传的功能,大部分时候能够满足我们的需要. 但有时候这个功 ...
- webpack4 系列教程(十四):Clean Plugin and Watch Mode
作者按:因为教程所示图片使用的是 github 仓库图片,网速过慢的朋友请移步<webpack4 系列教程(十四):Clean Plugin and Watch Mode>原文地址.更欢迎 ...
- RabbitMQ入门教程(十四):RabbitMQ单机集群搭建
原文:RabbitMQ入门教程(十四):RabbitMQ单机集群搭建 版权声明:本文为博主原创文章,遵循CC 4.0 BY-SA版权协议,转载请附上原文出处链接和本声明. 本文链接:https://b ...
- OCM_第十四天课程:Section6 —》数据库性能调优_各类索引 /调优工具使用/SQL 优化建议
注:本文为原著(其内容来自 腾科教育培训课堂).阅读本文注意事项如下: 1:所有文章的转载请标注本文出处. 2:本文非本人不得用于商业用途.违者将承当相应法律责任. 3:该系列文章目录列表: 一:&l ...
- WebGL简易教程(十四):阴影
目录 1. 概述 2. 示例 2.1. 着色器部分 2.1.1. 帧缓存着色器 2.1.2. 颜色缓存着色器 2.2. 绘制部分 2.2.1. 整体结构 2.2.2. 具体改动 3. 结果 4. 参考 ...
- 【转】机器学习教程 十四-利用tensorflow做手写数字识别
模式识别领域应用机器学习的场景非常多,手写识别就是其中一种,最简单的数字识别是一个多类分类问题,我们借这个多类分类问题来介绍一下google最新开源的tensorflow框架,后面深度学习的内容都会基 ...
- Redis教程(十四):内存优化介绍
转载于:http://www.itxuexiwang.com/a/shujukujishu/redis/2016/0216/142.html 一.特殊编码: 自从Redis 2.2之后,很多数据类型都 ...
- Unity3D脚本中文系列教程(十四)
http://dong2008hong.blog.163.com/blog/static/469688272014032134394/ WWWFrom 类Unity3D脚本中文系列教程(十三)辅助类. ...
- Spring Boot2 系列教程 (十四) | 统一异常处理
如题,今天介绍 SpringBoot 是如何统一处理全局异常的.SpringBoot 中的全局异常处理主要起作用的两个注解是 @ControllerAdvice 和 @ExceptionHandler ...
- Wix 安装部署教程(十四) -- 多语言安装包之用户许可协议
在上一篇中,留下了许可协议的问题,目前已经解决.感谢网友武全的指点! 问题 一般我们是用WixVariable 来设定许可协议.如下所示: <WixVariable Id="WixUI ...
随机推荐
- 通过axios实现数据请求
vue.js默认没有提供ajax功能的. 所以使用vue的时候,一般都会使用axios的插件来实现ajax与后端服务器的数据交互. 注意,axios本质上就是javascript的ajax封装,所以会 ...
- minio-搭建个人云存储服务
相信风靡全球的亚马逊 AWS S3 的存储云服务大家已经耳熟能详了,如何自己搭建一个私有的S3存储云服务呢?Minio 提供对象存储服务,兼容了 AWS S3 存储协议,用于非结构化的数据存.非结构化 ...
- 揭秘华为如此多成功项目的产品关键——Charter模板
很多推行IPD(集成产品开发)体系的公司在正式研发产品前,需要开发Charter,以确保产品研发方向的正确.Charter,即项目任务书或商业计划书.Charter的呈现标志着产品规划阶段的完成,能为 ...
- Yii框架Ar操作
1.$admin=Admin::model()->findAll($condition,$params); 该方法是根据一个条件查询一个集合,如: findAll("u ...
- NOIP模拟55
T1 Skip 解题思路 正解给的是线段树维护单调栈,但是我不会.. CDQ 维护斜率可做!!! 先得出一个朴素的 DP 方程:设 \(f_i\) 表示最后一场是 i 的最优解. 转移方程就是 \(f ...
- (性能测试)--记录一次高可用场景导致CPU资源升高
测试场景:高可用场景--限流测试: 被测交易:查询类交易,HTTP协议: 交易链路:jmeter - web - coimpre(前置服务) -- coimbp -- cobp (coimbp .co ...
- vmware vmnat1和vmnat8在真机网络适配器中消失
在真机的网络适配器中,发现只有两张网卡.缺少vmnat1和vmnat8 一,查看虚拟网络编辑器是否连接 二,如果没有连接,勾选连接就好了. 三,如果连接了,真机网络适配器仍然只有两张网络适配器. 1. ...
- 视图结构 wxml 列表渲染 for
WXML是框架设计的一套标签语言,结合基础组件.事件系统,可以构建出页面的结构. wxml是一个严格的标记性语言,有开始就必须有结束,单标签就一个有结束符 5.1.数据绑定 在js逻辑层中定义数据源, ...
- Java中可以用的大数据推荐算法
在Java中实现大数据推荐算法时,通常会使用一些开源的机器学习库,如Apache Mahout.Weka.DL4J(DeepLearning4j,用于深度学习)或者Spark MLlib(用于在Spa ...
- go 1.6 废弃 io/ioutil 包后的替换函数
go 1.6 废弃 io/ioutil 包后的替换函数 io/ioutil 替代 ioutil.ReadAll -> io.ReadAll ioutil.ReadFile -> os.R ...