用R包来下载sra数据
1)介绍
我们用SRAdb library来对SRA数据进行处理。 SRAdb 可以更方便更快的接入 metadata associated with submission, 包括study, sample, experiment, and run. SRAdb 包通过 NCBI SRA数据库中的metadata信息 作用. 首先dbConnect ()接入 R system 中的local database systems, 所有的搜索就在本地文件的基础上进行。
the queries we tried with the dbGetQuery function are passed in the form of SQL queries, which is a Select From Where framework. This part actually requires the
RSQLite package, which is installed when installing the SRAdb package, as a dependency. The getSRA function can actually do a full text search in the SRA data again via RSQLite and fetch the data in the selected fields for the query.
2)下载
source("http://bioconductor.org/biocLite.R")
biocLite("SRAdb")
library(SRAdb)
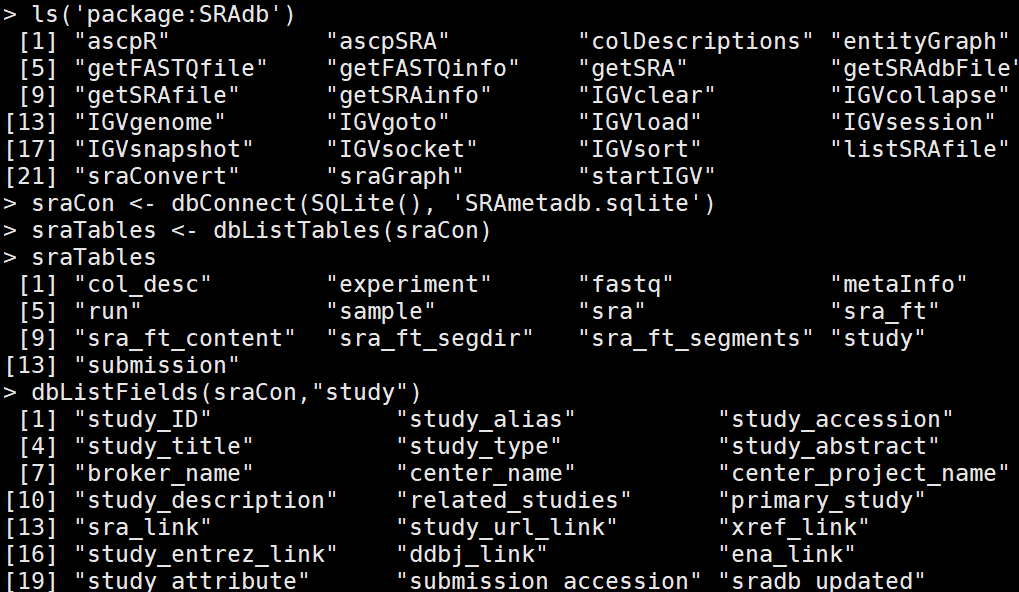
3)了解SRA database
#sqlFile <- getSRAdbFile() #在线获取,太大了,不要这样做。
sraCon <- dbConnect(SQLite(), 'SRAmetadb.sqlite') #于是我下载了这个文件,压缩文件2个G(解压后36个G),然后读取了这个文件,相当于下载nr库到本地。
sraTables <- dbListTables(sraCon) # investigate the content of the database
dbListFields(sraCon,"study")
#########关键词keyword: embryo
myHit <- dbGetQuery(sraCon, paste("select study_accession,study_title from study where","study_description like'%embryo'",sep=" ")) #
myHit <- getSRA( search_terms = "brain", out_types = c('run','study'), sraCon) #free text收索
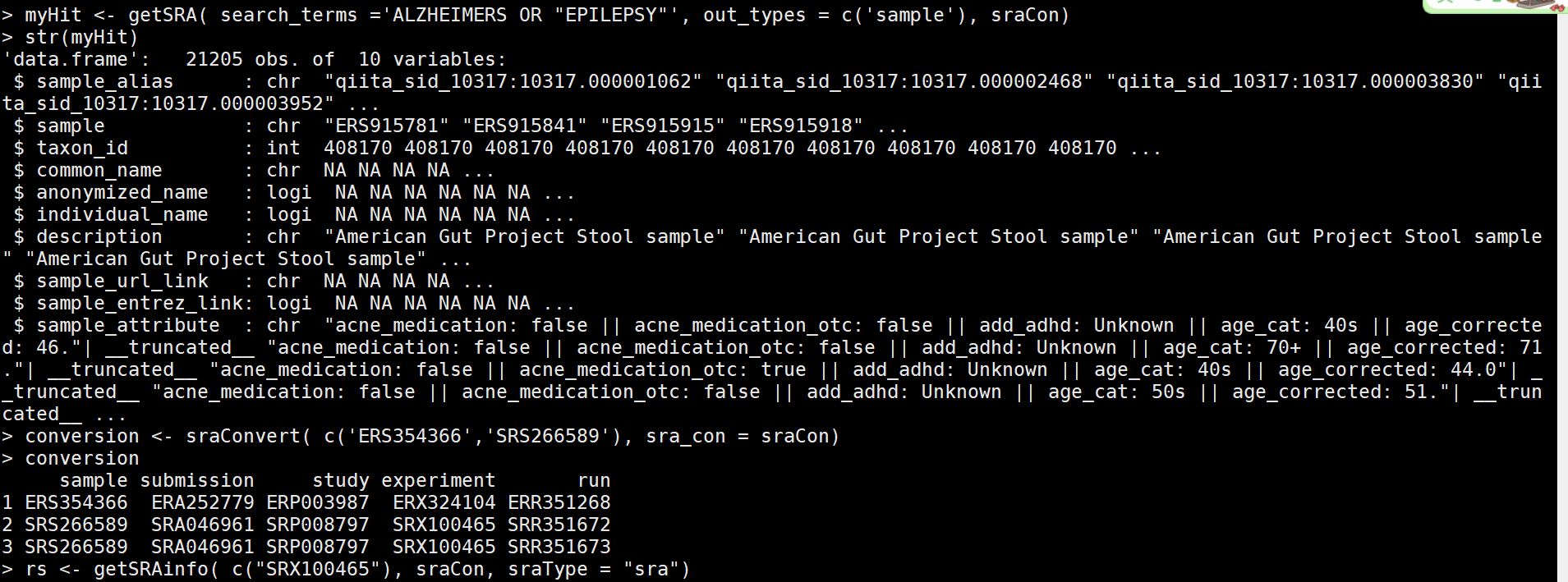
myHit <- getSRA( search_terms ='Alzheimers OR "EPILEPSY"', out_types = c('sample'), sraCon) #逻辑收索
4)从SRA database下载数据
myHit <- getSRA( search_terms ='ALZHEIMERS OR "EPILEPSY"', out_types = c('sample'), sraCon) #关键词收索
conversion <- sraConvert( c('ERS354366','SRS266589'), sra_con = sraCon) #选择其中的2个,查看信息
conversion
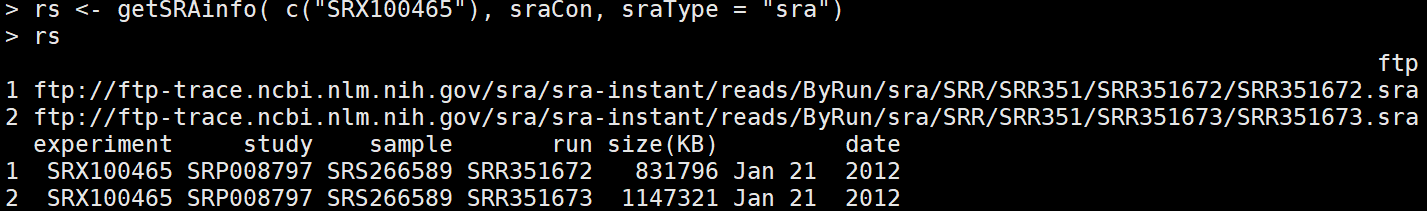
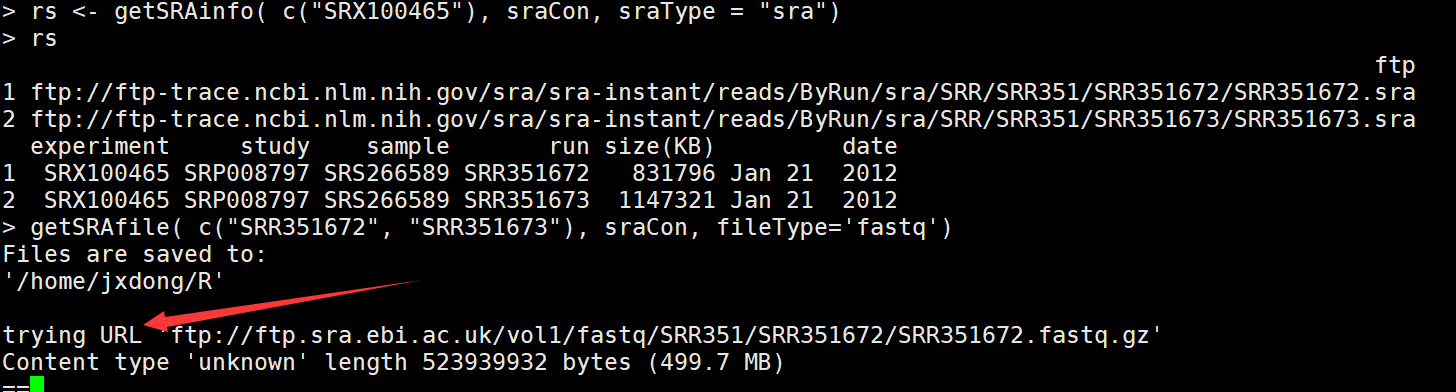
rs <- getSRAinfo( c("SRX100465"), sraCon, sraType = "sra") #选择其中一个看相应的信息,会显示出ftp地址
getSRAfile( c("SRR351672", "SRR351673"), sraCon, fileType='fastq') ##下载感兴趣的run
5)下载完fq文件后,用R进行读取
install.packages("R.utils")
library(R.utils) #下载数据用
download.file(url="ftp://ftp.ddbj.nig.ac.jp/ddbj_database/dra/fastq/SRA000/SRA000241/SRX000122/SRR000648.fastq.bz2", destfile = "SRR000648.fastq.bz2")
bunzip2(list.files(pattern = ".fastq.bz2$")) #解压
biocLite("ShortRead")
library(ShortRead) #读取fq文件
MyFastq <- readFastq(getwd(), pattern=".fastq") #小心运行,要至少8G内存
readLines("SRR000648.fastq", 4) # first four lines of the file
6)下载并读取比对数据(bam)
download.file(url="http://genome.ucsc.edu/goldenPath/help/examples/bamExample.bam", destfile = "bamExample.bam")
library(Rsamtools)
bam <- scanBam("bamExample.bam") #读取bam
names(bam[[1]]) #查看bam的信息
countBam("bamExample.bam") #统计bam信息 what <- c("rname", "strand", "pos", "qwidth", "seq") #只读取其中的几列
param <- ScanBamParam(what=what)
bam2 <- scanBam("bamExample.bam", param=param)
names(bam2[[1]])
bam_df <- do.call("DataFrame", bam[[1]]) # Read the data as a DataFrame object
head(bam_df) table(bam_df$rname == '21' & bam_df$flag == 16) #提取符合指定要求的sequences,即flag=16为reverse strands

7)对原始raw NGS data 的预处理
prefetch SRR000648
prefetch SRR000657
fastq-dump --split-3 -O ./ SRR000657
fastq-dump --split-3 -O ./ SRR000648
library(ShortRead)
myFiles <- list.files(getwd(), "fastq", full=TRUE)
myFQ <- lapply(myFiles, readFastq)
myQual <- FastqQuality(quality(quality(myFQ[[1]]))) #读取质量
readM <- as(myQual, "matrix") #将质控转化为矩阵
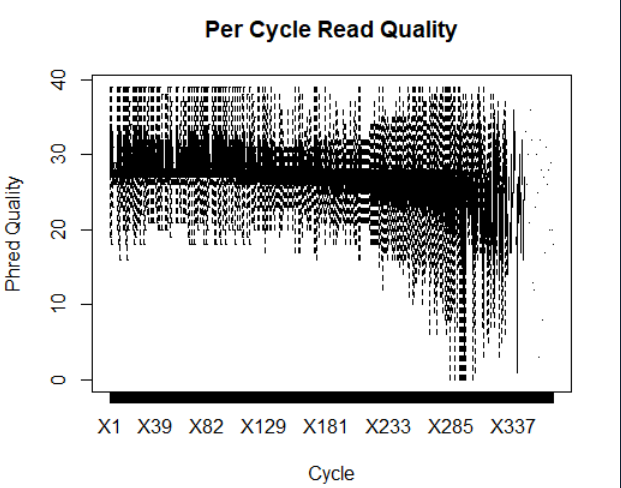
boxplot(data.frame(readM), outline = FALSE, main="Per Cycle Read Quality", xlab="Cycle", ylab="Phred Quality") #画箱型图
用R包来下载sra数据的更多相关文章
- NCBI下载sra数据(新)
今天要上NCBI下载sra数据发现没有下载的链接,网上查发现都是老的方法,NCBI页面已经变更,于是看了NCBI的help,并且记录下来新版的sra数据下载方法,要用NCBI的工具SRA Tool ...
- NCBI下载SRA数据
从NCBI下载数据本来是一件很简单的事情,但是今天碰到几个坑: 1.paper里没有提供SRA数据号.也没有提供路径: 2.不知道文件在ftp的地址,不能直接用wget下载 所以通过在NCBI官网,直 ...
- 资料:mnist.pkl.gz数据包的下载以及数据内容解释
deeplearning.net/data/mnist/mnist.pkl.gz The MNIST dataset consists of handwritten digit images and ...
- 从Github上轻松安装R包—githubinstall包--转载
1.综述 越来越多的R包正在由世界上不同的人所创建,其中一部分原因是devtools包使得开发R包1变得更加简单.devtools包不仅让开发R包变得简单,而且用于分发R包. 当开发者发布一个R包的时 ...
- 多组学分析及可视化R包
最近打算开始写一个多组学(包括宏基因组/16S/转录组/蛋白组/代谢组)关联分析的R包,避免重复造轮子,在开始之前随便在网上调研了下目前已有的R包工具,部分罗列如下: 1. mixOmics 应该是在 ...
- 查询、下载GWAS目录数据的R包(gwasrapidd)
目前GWAS方向发了很多文献,但是并没有一个很完善的R包对这些文献的数据进行汇总. 接下来推荐的这个是最新发表的GWAS数据汇总R包.看了一下功能齐全,但是数据不是收录的很齐全. 下面具体讲一下. ...
- R/Bioconductor包的下载和安装,升级
R包:基本包(自动加载)和推荐包(安装R时也会下载,但需要手动加载),拓展包(其他包,手动加载). 安装好的包将被放在一个指定的目录下.这个目录被称为库(Library).当需要使用到某一个包的时候, ...
- DT包 -- R语言中自定义表格数据
DT 包提供了 JavaScript 库 DataTables 的一个R接口,它使得R对象(矩阵或数据框)可以在HTML页面上显示为表格. 该包的DataTables函数生成的表格提供了数据的筛选.分 ...
- 使用GEOquery下载GEO数据--转载
最近需要下载一大批GEO上的数据,问题是我要下载的Methylation数据根本就没有sra文件,换言之不能使用Aspera之类的数据进行下载.但是后来我发现了GEOquery这个不错的R包,不知道是 ...
随机推荐
- 异步Socket服务器与客户端
本文灵感来自Andre Azevedo 在CodeProject上面的一片文章,An Asynchronous Socket Server and Client,讲的是异步的Socket通信. S ...
- C# Web Service 初级教学
原文连接:http://www.codeproject.com/cs/webservices/myservice.asp作者:Chris Maunder Introduction Creating y ...
- 开始转型学习java
什么编程语言这些都是一样的,编程思想都是一样的.只不过是表现形式. 标识符 每一个字符在ascll码表例都有对应的数字 所以字符和数字是可以相加的 'a'+1 也可以显示数字对应的字符 (ch ...
- spring Annotation based configuration
spring 注解相关 https://docs.spring.io/spring/docs/3.0.0.M3/reference/html/ch04s11.html
- ALGO-2_蓝桥杯_算法训练_最大最小公倍数
问题描述 已知一个正整数N,问从1~N中任选出三个数,他们的最小公倍数最大可以为多少. 输入格式 输入一个正整数N. 输出格式 输出一个整数,表示你找到的最小公倍数. 样例输入 样例输出 数据规模与约 ...
- 导入wordpress数据库到mysql报错
mysql字符集编码错误的导入数据会提示错误了,这个和插入数据一样如果保存的数据与mysql编码不一样那么肯定会出现导入乱码或插入数据丢失的问题,下面我们一起来看一个例子. 恢复数据库报错:由于字符集 ...
- bzoj3668 起床困难综合症
Description 21 世纪,许多人得了一种奇怪的病:起床困难综合症,其临床表现为:起床难,起床后精神不佳.作为一名青春阳光好少年,atm 一直坚持与起床困难综合症作斗争.通过研究相关文献,他找 ...
- hdu 1966 Pie
Pie Time Limit: 5000/1000 MS (Java/Others) Memory Limit: 65536/32768 K (Java/Others)Total Submiss ...
- Executor框架(一)Executor框架介绍
Executor框架简介 Executor框架的两级调度模型 在HotSpot VM的线程模型中,Java线程被一对一映射为本地操作系统线程.Java线程启动时会创建一个本地操作系统线程:当Jav ...
- ORM sqlachemy学习
内容: 1.ORM介绍 2.SQLAlchemy介绍 3.SQLAlchemy内部处理 4.SQLAlchemy使用 参考: http://www.cnblogs.com/wupeiqi/articl ...