Directional RNA-seq data -which parameters to choose?
Directional RNA-seq data -which parameters to choose?
REF:
https://chipster.csc.fi/manual/library-type-summary.html
Directional RNA-seq methods are gaining popularity. Several protocols and products are available for the library preparation step, and different tools and softwares have different options to take these into account. Since this has caused a lot of confusion due to incoherent parameter naming, we try to clarify this issue a bit here.
To be able to select the right parameters for your data, first you need to know which library prep method was used when generating your data. In general, there are three types of library preps:
- un-stranded
- "second-strand" = directional, where the first read of the read pair (or in case of single end reads, the only read) is from the transcript strand
- "first-strand" = directional, where the first read (or the only read in case of SE) is from the opposite strand.
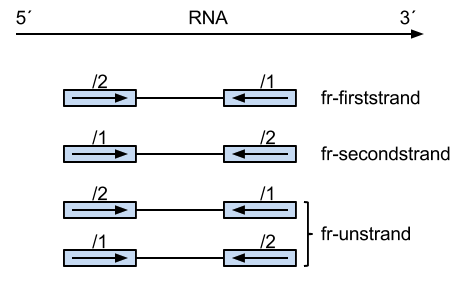
Summary of library type protocols
(borrowed from:
http://onetipperday.sterding.com/2012/07/how-to-tell-which-library-type-to-use.html)
The reads on the left are from the same strand as the transcript,
and their pairs on the right are from the opposing strand.
The number above the read states which read it is, the first (/1) or the
second (/2). Thus, perhaps a bit unintuitively, the first case,
"fr-firststrand" is such that the first read (/1) is actually from the
opposing strand as the transcript, and second read (/2) is from the
transcript strand.
Why is this so important? If you use wrong directionality parameter, in the read counting step
the reads are considered to be from the wrong strand. This means that in the cases where there
are no genes on that other strand, you won't get any hits, and if there are genes in the same
location on the other strand, your reads are counted for that wrong gene.
How can I check I chose correctly? It's a good idea to check that!
Using the tool RNA-seq strandedness inference and inner distance estimation using RseQC:
We added this tool under the Quality control
category to help you. The tool aligns subsets of the input FASTQ files
against the reference genome,
and this alignment is then compared to the reference annotation to
deduce the strandedness. Make sure you select the correct reference when
running the tool.
Check out the
help page of this tool for more information!
In aligners like HISAT2 and Tophat you can also do a comparison and check the mapping rate. Take a small subset of your reads and
run HISAT2/TopHat with the different parameters and compare the results, and check the log file.
In HTSeq you can also run the tool with different options and
check the number of reads that are not counted for any gene (=the
"no-feature reads").
(In Chipster, open file htseq-count-info.txt).
Be extra careful to assign the paired files correctly! Using these parameters assumes you are giving the
files in specific order: read1, read2. In Chipster always check from the parameters window that your
files are assigned correctly.
Below we list some common library preparation kits and their corresponding parameters
in different tools. Is your kit missing from the list? If you have the data generated with that kit and figure
out the library type, please let us know too, so we can add that kit to the list below.
Unstranded:
Information regarding the strand is not conserved
(it is lost during the amplification of the mRNA fragments).
Kits:
TruSeq RNA Sample Prep kit
Parameters:
HISAT2 / TopHat / Cufflinks / Cuffdiff: library-type fr-unstranded
HTSeq: stranded -- no
Directional, first strand:
The second read (read 2) is from the original RNA strand/template, first read (read 1) is from the
opposite strand.
The information of the strand is preserved as the original RNA strand is degradated due to the
dUTPs incorporated in the second synthesis step.
Kits:
All dUTP methods, NSR, NNSR
TruSeq Stranded Total RNA Sample Prep Kit
TruSeq Stranded mRNA Sample Prep Kit
NEB Ultra Directional RNA Library Prep Kit
Agilent SureSelect Strand-Specific
Parameters:
HISAT2 / TopHat / Cufflinks / Cuffdiff: library-type fr-firststrand
HTSeq: stranded -- reverse
Directional, second strand:
The first read (read 1) is from the original RNA strand/template, second read (read 2) is from the
opposite strand.
The directionality is preserved, as different adapters are ligated to different ends of the fragment.
Kits:
Directional Illumina (Ligation), Standard SOLiD
ScriptSeq v2 RNA-Seq Library Preparation Kit
SMARTer Stranded Total RNA
Encore Complete RNA-Seq Library Systems
NuGEN SoLo
Parameters:
HISAT2 / TopHat / Cufflinks / Cuffdiff: library-type fr-secondstrand
HTSeq: stranded -- yes
Note also that the --fr/--rf/--ff or "Order of mates to align" parameter in Bowtie has similar
sounding parameter options: [--fr: "Forward/reverse", --rf: "Reverse/Forward", --ff: "Forward/forward"].
However, these parameters are a bit different story, as they explain how the paired end reads are
oriented towards each other (-> <-, -> -> or <- ->). The default (--fr, -> <-) is appropriate for
Illumina's paired-end reads: it means that read 1 appears upstream of the reverse complement of read 2,
or vice versa. When running TopHat, the library-type parameter is delivered to Bowtie, so the
user doesn't have to worry about that too much.
================
I think the issue here is that the R and F options yield the same results. We just encountered this same issue with PE sequencing (RF give the same results as FR).
ref:
https://github.com/infphilo/hisat2/issues/61
================
--fr/--rf/--ff should rarely be set, since they refer to the relative orientation of reads and basically everything these days is --fr. Mate-pairs were --rf, but you don't see those much any more.
--rna-strandedness is a very different option, since it sets how reads are expected to align against genes. Most stranded protocols in use these days follow the dUTP-method, where read #2 in a pair has the same orientation as the transcript from which it arose. So either R or RF would typically be appropriate, unless the library is unstranded. In practice, I expect this is more useful if you plan to run stringTie downstream, since then the XS auxiliary tag is set appropriately.
ref:
https://bioinformatics.stackexchange.com/questions/4074/hisat2-which-option-should-mention-for-strand-specific-library-read
================
Thanks a lot for your always useful comments. I thought it was so, but then I ran into a different problem. I first aligned with the option forward --fr, and got "70% reads aligned concordantly exactly 1 time". It seemed OK. Then, noticing my mistake with the library type, ran the same fastqsanger input files with option forward --rf and got about 98% "aligned concordantly 0 times". I still cannot explain what happened. Any idea? Both times I did input first read1 then read2 on the tool form.
ref:
https://biostar.usegalaxy.org/p/27320/
================
I have a pair end strand-specific library type: fr-secondstrand so could you suggest for Hisat2 it should be --fr?
There are two option in hisat2 : --rna-strandness and --fr/--rf/--ff . please suggest which option should I mention from these two.
--rna-strandness fr
-- fr
ref:
https://github.com/infphilo/hisat2/issues/109
================
I'm working with RNA-seq data. For the analysis I will be using HISAT2 to align sequencing reads to the human genome GRCh38. Samples are subjected to strand-specific RNA sequencing using poly-A selection protocol and sequenced on the Illumina HiSeq 2000 system. They are paired-end reads.
I would like to know how the Hisat2 command need to be given for strand-specific option.
--rna-strandness should be RF or FR
For Hisat2 there is two things to take care of:
If its single ended data and forward stranded you need to set: -rna-strandness F If its paired end data and forward stranded you need to set: -rna-strandness FR
Similar, if its reverse stranded for SE data: -rna-strandness R or (Paired End, reverse stranded) -rna-strandness RF
You may have to check in more detail what the protocol actually does.
----------------
Is there a possibility to know whether it is forward stranded or reverse stranded from fatsq files I have (sample1_1.fastq, sample1_2.fatsq) ?
--------------
If you don't know if for sure, you can try using RSeQC to infer the strandedness:
-----------------
Most are fr-firststrand (dUTP based methods). But yes it's good to check with infer_experiment.py from RSeQC. For fr-firststrand you should use RF.
ref:
https://www.biostars.org/p/297399/
================
I also applied --rna-strandness F and --rna-strandness R respectively (with --tmo option on). My reads should be F. However, F and R gave me exactly the same output, with only diffice at " XS:A:" - one is +, the other gives - for a same read.
With this option being used, every read alignment will have an XS attribute tag: '+' means a read belongs to a transcript on '+' strand of genome. '-' means a read belongs to a transcript on '-' strand of genome.
ref:
https://www.biostars.org/p/307073/
================
RF vs FR
| Library Kit | Stranded | 5p to 3p IGV | TopHat (--library-type parameter) | HISAT2 (--rna-strandness) | HTSeq (--stranded/-s) | Picard (STRAND_SPECIFICITY option of CollectRnaSeqMetrics) | Kallisto quant |
|---|---|---|---|---|---|---|---|
| TruSeq Strand Specific Total RNA | Yes | F2R1 | fr-firststrand | R/RF | reverse | SECOND_READ_TRANSCRIPTION_STRAND | --fr-stranded |
| NuGEN Encore | Yes | F1R2 | fr-secondstrand | F/FR | yes | FIRST_READ_TRANSCRIPTION_STRAND | --rf-stranded |
| NuGEN OvationV2 | No | F2R1 or F1R2 | fr-unstranded | NONE | no | NONE | NONE |
REF:
https://www.biostars.org/p/169942/
https://www.biostars.org/p/243877/
https://github.com/griffithlab/rnaseq_tutorial/blob/master/manuscript/supplementary_tables/supplementary_table_5.md
Directional RNA-seq data -which parameters to choose?的更多相关文章
- RNA seq 两种计算基因表达量方法
两种RNA seq的基因表达量计算方法: 1. RPKM:http://www.plob.org/2011/10/24/294.html 2. RSEM:这个是TCGAdata中使用的.RSEM据说比 ...
- RNA -seq
RNA -seq RNA-seq目的.用处::可以帮助我们了解,各种比较条件下,所有基因的表达情况的差异. 比如:正常组织和肿瘤组织的之间的差异:检测药物治疗前后,基因表达的差异:检测发育过程中,不同 ...
- Linux top、VIRT、RES、SHR、SWAP(S)、DATA Memory Parameters Detailed
catalog . Linux TOP指令 . VIRT -- Virtual Image (KB) . RES -- Resident size (KB) . SHR -- Shared Memor ...
- An Edge-Guided Image Interpolation Algorithm via Directional Filtering and Data Fusion【翻译】
基于定向滤波和数据融合的边缘引导图像插值算法 http://ieeexplore.ieee.org/document/1658087/ 摘要: 保留边缘结构对于从低分辨率对应物重建高分辨率图像的图像插 ...
- RNA测序相对基因表达芯片有什么优势?
RNA测序相对基因表达芯片有什么优势? RNA-Seq和基因表达芯片相比,哪种方法更有优势?关键看适用不适用.那么RNA-Seq适用哪些研究方向?是否您的研究?来跟随本文了解一下RNA测序相对基因表达 ...
- SQL to JSON Data Modeling with Hackolade
Review: SQL to JSON data modeling First, let’s review, the main way to represent relations in a rela ...
- RNA测序的质量控制
RNA测序的质量控制 发表评论 3,112 A+ 所属分类:Transcriptomics 收 藏 ENCODE项目向我们揭示,人类基因组中超过70%能得到转录,只不过不会发生在同一个细胞里.为 ...
- [Windows Azure] Data Management and Business Analytics
http://www.windowsazure.com/en-us/develop/net/fundamentals/cloud-storage/ Managing and analyzing dat ...
- Linux command line exercises for NGS data processing
by Umer Zeeshan Ijaz The purpose of this tutorial is to introduce students to the frequently used to ...
随机推荐
- 《大话设计模式》c++实现 工厂模式
工厂模式 工厂模式(Factory Pattern)是 Java 中最常用的设计模式之一.这种类型的设计模式属于创建型模式,它提供了一种创建对象的最佳方式. 在工厂模式中,我们在创建对象时不会对客户端 ...
- python windows 下pip easy_install 使用错误的问题
最近电脑重装了系统,又重新安装python .在官网下载了安装包后电脑成功安装了,但使用pip命令时出现以下错误 Fatal error in launcher: Unable to create p ...
- python pandas模块,nba数据处理(1)
pandas提供了使我们能够快速便捷地处理结构化数据的大量数据结构和函数.pandas兼具Numpy高性能的数组计算功能以及电子表格和关系型数据(如SQL)灵活的数据处理能力.它提供了复杂精细的索引功 ...
- Python全栈-day10-函数2
函数高级篇 1.函数嵌套 1)嵌套定义 在函数内定义另外一个函数 def func(): print('嵌套定义') def func1(): print('这是一个嵌套函数') def func2( ...
- html5-table布局
<!DOCTYPE html><html lang="en"><head> <meta charset="UTF-8&qu ...
- Game (思维)
#include<bits/stdc++.h> using namespace std; ; char str[maxn][maxn]; int cntx[maxn], cnty[maxn ...
- Linux基础命令---修改用户密码
passwd 更改用户密码,超级用户可以修改所有用户密码,普通用户只能修改自己的密码.这个任务是通过调用LinuxPAM和LibuserAPI来完成的.本质上,它使用LinuxPAM将自己初始化为一个 ...
- mongodb安全权限设定
mongodb安全权限设定 如何防范此类攻击? 做好访问认证.打开你的MongoDB配置文件(.conf),设置为auth=true 做好防火墙设置.建议管理者关闭27017端口的访问. Bind_i ...
- 浅析PAC,修改PAC文件及user-rule文件实现自动代理
浅析PAC,修改PAC文件及user-rule文件实现自动代理 代理自动配置(英语:Proxy auto-config,简称PAC)是一种网页浏览器技术,用于定义浏览器该如何自动选择适当的代理服务器来 ...
- 使用Fiddler测试WebApi接口
Fiddler是好用的WebApi调试工具之一,它能记录所有客户端和服务器的http和https请求,允许你监视,设置断点,甚至修改输入输出数据,Fiddler 是以代理web服务器的形式工作的,使用 ...