单细胞数据初步处理 | drop-seq | QC | 质控 | 正则化 normalization
比对
The raw Drop-seq data was processed with the standard pipeline (Drop-seq tools version 1.12 from McCarroll laboratory). Reads were aligned to the ENSEMBL release 84Mus musculusgenome.
10x Genomics data was processed using the same pipeline as for Drop-seq data, adjusting the barcode locations accordingly
我还没有深入接触10x和drop-seq的数据,目前的10x数据都是用官网cellranger跑出来的。
质控
We selected cells for downstream processing in each Drop-seq run, using the quality control metrics output by the Drop-seq tools package9, as well as metrics derived from the UMI matrix.
1) We first removed cells with a low number (<700) of unique detected genes. From the remaining cells, we filtered additional outliers.
2) We removed cells for which the overall alignment rate was less than the mean minus three standard deviations.
3) We removed cells for which the total number of reads (after log10 transformation) was not within three standard deviations of the mean.
4) We removed cells for which the total number of unique molecules (UMIs, after log10 transformation) was not within three standard deviations of the mean.
5) We removed cells for which the transcriptomic alignment rate (defined by PCT_USABLE_BASES) was not within three standard deviations of the mean.
6) We removed cells that showed an unusually high or low number of UMIs given their number of reads by fitting a loess curve (span= 0.5, degree= 2) to the number of UMIs with number of reads as predictor (both after log10 transformation). Cells with a residual more than three standard deviations away from the mean were removed.
7) With the same criteria, we removed cells that showed an unusually high or low number of genes given their number of UMIs. Of these filter steps, step 1 removed the majority of cells.
Steps 2 to 7 removed only a small number of additional cells from each eminence (2% to 4%), and these cells did not exhibit unique or biologically informative patterns of gene expression.
1. 过滤掉基因数量太少的细胞;
2. 过滤基因组比对太差的细胞;
3. 过滤掉总reads数太少的细胞;
4. 过滤掉UMI太少的细胞;
5. 过滤掉转录本比对太少的细胞;
6. 根据统计分析,过滤reads过多或过少的细胞;
7. 根据统计分析,过滤UMI过低或过高的细胞;
注:连过滤都有点统计的门槛,其实也简单,应该是默认为正态分布,去掉了左右极端值。
还有一个就是简单的拟合回归,LOESS Curve Fitting (Local Polynomial Regression)
How to fit a smooth curve to my data in R?
正则化
The raw data per Drop-seq run is a UMI count matrix with genes as rows and cells as columns. The values represent the number of UMIs that were detected. The aim of normalization is to make these numbers comparable between cells by removing the effect of sequencing depth and biological sources of heterogeneity that may confound the signal of interest, in our case cell cycle stage.
目前有很多正则化的方法,但是作者还是自己开发了一个。
正则化就是去掉一些影响因素,使得我们的数据之间可以相互比较。这里就提到了两个最主要的因素:测序深度和细胞周期。
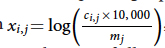
A common approach to correct for sequencing depth is to create a new normalized expression matrix x with (see Fig), in which ci,j is the molecule count of gene i in cell j and mj is the sum of all molecule counts for cell j. This approach assumes that ci,j increases linearly with mj, which is true only when the set of genes detected in each cell is roughly the same.
可以看到常规的正则化方法是不适合的,
However, for Drop-seq, in which the number of UMIs is low per cell compared to the number of genes present, the set of genes detected per cell can be quite different. Hence, we normalize the expression of each gene separately by modelling the UMI counts as coming from a generalized linear model with negative binomial distribution, the mean of which can be dependent on technical factors related to sequencing depth. Specifically, for every gene we model the expected value of UMI counts as a function of the total number of reads assigned to that cell, and the number of UMIs per detected gene (sum of UMI divided by number of unique detected genes).
这个就有些门槛了,用了广义线性回归模型来做正则化。
To solve the regression problem, we use a generalized linear model (glm function of base R package) with a regularized overdispersion parameter theta. Regularizing theta helps us to avoid overfitting which could occur for genes whose variability is mostly driven by biological processes rather than sampling noise and dropout events. To learn a regularized theta for every gene, we perform the following procedure.
1) For every gene, obtain an empirical theta using the maximum likelihood model (theta.ml function of the MASS R package) and the estimated mean vector that is obtained by a generalized linear model with Poisson error distribution.
2) Fit a line (loess, span = 0.33, degree = 2) through the variance–mean UMI count relationship (both log10 transformed) and predict regularized theta using the fit. The relationship between variance and theta and mean is given by variance= mean + (mean2/theta).
Normalized expression is then defined as the Pearson residual of the regression model, which can be interpreted as the number of standard deviations by which an observed UMI count was higher or lower than its expected value. Unless stated otherwise, we clip expression to the range [-30, 30] to prevent outliers from dominating downstream analyses.
好的是,代码人家都给出来了,你去跑跑,就能猜出大致的意思。
# for normalization
# regularized overdispersion parameter theta. Regularizing theta helps us to avoid overfitting which could occur for genes whose variability is mostly driven by biological processes rather than sampling noise and dropout events.
# divide all genes into 64 bins
theta.reg <- function(cm, regressors, min.theta=0.01, bins=64) {
b.id <- (1:nrow(cm)) %% max(1, bins, na.rm=TRUE) + 1
cat(sprintf('get regularized theta estimate for %d genes and %d cells\n', nrow(cm), ncol(cm)))
cat(sprintf('processing %d bins with ca %d genes in each\n', bins, round(nrow(cm)/bins, 0)))
theta.estimate <- rep(NA, nrow(cm))
# For every gene, obtain an empirical theta using the maximum likelihood model (theta.ml function of the MASS R package)
for (bin in sort(unique(b.id))) {
sel.g <- which(b.id == bin)
bin.theta.estimate <- unlist(mclapply(sel.g, function(i) {
# estimated mean vector that is obtained by a generalized linear model with Poisson error distribution
as.numeric(theta.ml(cm[i, ], glm(cm[i, ] ~ ., data = regressors, family=poisson)$fitted))
}), use.names = FALSE)
theta.estimate[sel.g] <- bin.theta.estimate
cat(sprintf('%d ', bin))
}
cat('done\n')
raw.mean <- apply(cm, 1, mean)
log.raw.mean <- log10(raw.mean)
var.estimate <- raw.mean + raw.mean^2/theta.estimate # Fit a line (loess, span = 0.33, degree = 2) through the variance–mean UMI count relationship (both log10 transformed)
fit <- loess(log10(var.estimate) ~ log.raw.mean, span=0.33)
# predict regularized theta using the fit. The relationship between variance and theta and mean is given by variance= mean + (mean2/theta)
theta.fit <- raw.mean^2 / (10^fit$fitted - raw.mean) to.fix <- theta.fit <= min.theta | is.infinite(theta.fit)
if (any(to.fix)) {
cat('Fitted theta below', min.theta, 'for', sum(to.fix), 'genes, setting them to', min.theta, '\n')
theta.fit[to.fix] <- min.theta
}
names(theta.fit) <- rownames(cm)
return(theta.fit)
} nb.residuals.glm <- function(y, regression.mat, fitted.theta, gene) {
fit <- 0
try(fit <- glm(y ~ ., data = regression.mat, family=negative.binomial(theta=fitted.theta)), silent=TRUE)
if (class(fit)[1] == 'numeric') {
message(sprintf('glm and family=negative.binomial(theta=%f) failed for gene %s; falling back to scale(log10(y+1))',
fitted.theta, gene))
return(scale(log10(y+1))[, 1])
}
return(residuals(fit, type='pearson'))
} ## Main function
norm.nb.reg <- function(cm, regressors, min.theta=0.01, bins=64, theta.fit=NA, pr.th=NA, save.theta.fit=c()) {
cat('Normalizing data using regularized NB regression\n')
cat('explanatory variables:', colnames(regressors), '\n')
if (any(is.na(theta.fit))) {
theta.fit <- theta.reg(cm, regressors, min.theta, bins)
if (is.character(save.theta.fit)) {
save(theta.fit, file=save.theta.fit)
}
} b.id <- (1:nrow(cm)) %% max(1, bins, na.rm=TRUE) + 1
cat('Running NB regression\n')
res <- matrix(NA, nrow(cm), ncol(cm), dimnames=dimnames(cm))
for (bin in sort(unique(b.id))) {
sel.g <- rownames(cm)[b.id == bin]
expr.lst <- mclapply(sel.g, function(gene) nb.residuals.glm(cm[gene, ], regressors, theta.fit[gene], gene), mc.preschedule = TRUE)
# Normalized expression is then defined as the Pearson residual of the regression model, which can be interpreted as the number of standard deviations by which an observed UMI count was higher or lower than its expected value.
res[sel.g, ] <- do.call(rbind, expr.lst)
cat(sprintf('%d ', bin))
}
cat('done\n')
# clip expression to the range [-30, 30] to prevent outliers from dominating downstream analyses
if (!any(is.na(pr.th))) {
res[res > pr.th] <- pr.th
res[res < -pr.th] <- -pr.th
}
attr(res, 'theta.fit') <- theta.fit
return(res)
}
单细胞数据初步处理 | drop-seq | QC | 质控 | 正则化 normalization的更多相关文章
- Python数据预处理—归一化,标准化,正则化
关于数据预处理的几个概念 归一化 (Normalization): 属性缩放到一个指定的最大和最小值(通常是1-0)之间,这可以通过preprocessing.MinMaxScaler类实现. 常用的 ...
- 单细胞数据整合方法 | Comprehensive Integration of Single-Cell Data
操作代码:https://satijalab.org/seurat/ 依赖的算法 CCA CANONICAL CORRELATION ANALYSIS | R DATA ANALYSIS EXAMPL ...
- 深度挖坑:从数据角度看人脸识别中Feature Normalization,Weight Normalization以及Triplet的作用
深度挖坑:从数据角度看人脸识别中Feature Normalization,Weight Normalization以及Triplet的作用 周翼南 北京大学 工学硕士 373 人赞同了该文章 基于深 ...
- [转] sql 删除表数据的drop、truncate和delete用法
删除表数据的关键字,大家记得最多的可能就是delete.然而,我们做数据库开发,读取数据库数据.对另外的drop.truncate用得就比较少了. 1 drop 出没场合:drop table ta ...
- 单细胞数据高级分析之构建成熟路径 | Identifying a maturation trajectory
其实就是另一种形式的打分. 个人点评这种方法: 这篇文章发表在nature上,有点奇怪,个人感觉创新性和重要性还不够格,工具很多,但是本文基本都是自己开发的算法(毕竟satji就是搞统计出身的). 但 ...
- 单细胞数据高级分析之初步降维和聚类 | Dimensionality reduction | Clustering
个人的一些碎碎念: 聚类,直觉就能想到kmeans聚类,另外还有一个hierarchical clustering,但是单细胞里面都用得不多,为什么?印象中只有一个scoring model是用kme ...
- 单细胞数据高级分析之消除细胞周期因素 | Removal of cell cycle effect
The normalization method described above aims to reduce the effect of technical factors in scRNA-seq ...
- 单细胞数据normalization方法 | SCTransform
SCTransform Normalization and variance stabilization of single-cell RNA-seq data using regularized n ...
- QC学习三:Excel数据导入导出QC操作流程
环境: QC9 WindowsXP Office2007 1. 准备 1.通过Excel导入QC,需要下载Microsoft Excel Add-in: http://update.externa ...
随机推荐
- KNN(K-Nearest Neighbor)介绍
KNN(K-Nearest Neighbor)介绍 原文地址:https://www.cnblogs.com/nucdy/p/6349172.html Wikipedia上的 KNN词条 中有一个比较 ...
- 当模版引擎遇到点("."),会按照下列顺序查询:
字典查询,例如:foo["bar"] 属性或方法查询,例如:foo.bar 数字索引查询,例如:foo[bar]
- 从客户端(XXX)中检测到有潜在危险的Request.Form 值
aspx 页面出现 [HttpRequestValidationException (0x80004005):从客户端(TextBox1="<?xml version="1. ...
- vue学习【第三篇】:vue之node.js的简单介绍
什么是node.js 它是可以运行JavaScript的服务平台,可以吧它当做一门后端程序,只是它的开发语言是JavaScript 安装node.js node.js的特性 - 非阻塞IO模型 - 时 ...
- 闪存中的NorFlash、NandFlash及eMMC三者的区别【转】
本文转载自:https://blog.csdn.net/Blazar/article/details/77843655 快闪存储器(英语:Flash Memory),是一种电子式可清除程序化只读存储器 ...
- ubuntu安装微软雅黑和Consolas字体
原文:http://fooler5.iteye.com/blog/2406227 [字体下载] YaHeiConsolas.tar:http://www.mycode.net.cn/wp-conten ...
- 三星sm865
目录 样张 SSD-Z: CrystalDiskInfo: CrystalDiskMark: AS-SSD Benchmark: 颗粒检查: 扇区信息: HD Tune Pro: 三星Magician ...
- 微服务架构与实践4_Docker
构建Docker映像 定义Dockerfile=>Docker根据Dockerfile构建出映像 包含: 基础映像(父映像)信息 维护者信息 映像操作命令 容器启动命令 .net standar ...
- 数据结构和算法with Python
http://www.math.pku.edu.cn/teachers/qiuzy/ds_python/courseware/index.htm
- 5、lvs使用进阶(01)
四层.七层负载均衡的区别 https://jaminzhang.github.io/lb/L4-L7-Load-Balancer-Difference/ netfilter/iptables简介 ...