edgeR
1)简介
edgeR作用对象是count文件,rows 代表基因,行代表文库,count代表的是比对到每个基因的reads数目。它主要关注的是差异表达分析,而不是定量基因表达水平。
edgeR works on a table of integer read counts, with rows corresponding to genes and columns to independent libraries. The counts represent the total number of reads aligning to each gene (or other genomic locus).edgeR is concerned with differential expression analysis rather than with the quantification of expression levels. It is concerned with relative changes in expression levels between conditions,but not directly with estimating absolute expression levels.
edgeR作用的是真实的比对统计,因此不建议用预测的转录本
Note that edgeR is designed to work with actual read counts. We not recommend that predicted transcript abundances are input the edgeR in place of actual counts.
归一化原因:
技术原因影响差异表达分析:
1)Sequencing depth:统计测序深度(即代表的是library size);
2)RNA composition:个别异常高表达基因导致其它基因采样不足
3)GC content: sample-specific effects for GC-content can be detected
4)sample-specific effects for gene length have been detected
注意:edgeR必须是原始表达量,而不能是rpkm等矫正过的。
Note that normalization in edgeR is model-based, and the original read counts are not themselves transformed. This means that users should not transform the read counts in any way before inputing them to edgeR. For example, users should not enter RPKM or FPKM values to edgeR in place of read counts. Such quantities will prevent edgeR from correctly estimating the mean-variance relationship in the data, which is a crucial to the statistical strategies underlying edgeR.Similarly, users should not add artificial values to the counts before inputing them to edgeR.
2)安装
if("edgeR" %in% rownames(installed.packages()) == FALSE) {source("http://bioconductor.org/biocLite.R");biocLite("edgeR")}
suppressMessages(library(edgeR))
ls('package:edgeR')
3)矩阵构建及差异分析
需要构建2个矩阵:1、表达矩阵;2、分组矩阵( 实验设计);
-------------------------------------------------------表达矩阵-----------------------------------------
3.1、读取表达矩阵文件(Reading in the data)
#读取文件
rawdata <- read.delim("E:/software/R/R-3.5.0/library/edgeR/Meta/TableS1.txt", check.names=FALSE, stringsAsFactors=FALSE)
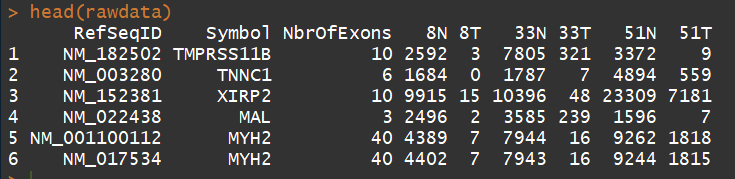
head(rawdata)
3.2 、构建DGEList对象
这里因为已经有rawdata的count文件,因此直接用DGEList()函数就行了,否则要用readDGE()函数
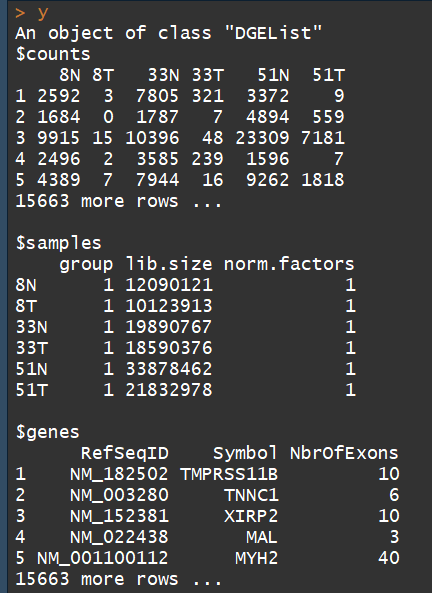
y <- DGEList(counts=rawdata[,4:9], genes=rawdata[,1:3])##构建DGEList对象
DGEList对象主要有三部分:
1、counts矩阵:包含的是整数counts;
2、samples数据框:包含的是文库(sample)信息。包含 lib.size列 :for the library size (sequencing depth) for each sample,如果不自定义, the library sizes will be computed from the column sums of the counts。其中还有一个group列,用于指定每个sample组信息
3、一个可选的数据框genes:gene的注释信息
3.3)数据注释( Annotation)
这里主要是因为该文章数据是前好多年的,因此需要过滤,symbol更新等。
1)The study was undertaken a few years ago, so not all of the RefSeq IDs provided by match RefSeq IDs currently in use. We retain only those transcripts with IDs in the current NCBI annotation, which is provided by the org.HS.eg.db package
2)因为edgeR默认使用NCBI中refSeq的ID,所以通过refseq Id 找到entrezID,然后通过entrezID对symbol更新
#######retain only those transcripts with IDs in the current NCBI annotation provided by the org.HS.eg.db######
library(org.Hs.eg.db)
idfound <- y$genes$RefSeqID %in% mappedRkeys(org.Hs.egREFSEQ)
y <- y[idfound,]
dim(y) ##15550 6
###################### 在注释中加入 Entrez Gene IDs #########################
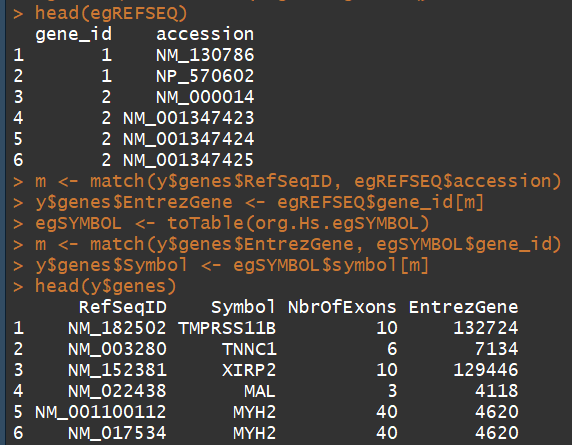
egREFSEQ <- toTable(org.Hs.egREFSEQ)
m <- match(y$genes$RefSeqID, egREFSEQ$accession)
y$genes$EntrezGene <- egREFSEQ$gene_id[m]
#####################用Entrez Gene IDs更新gene symbols##########################
egSYMBOL <- toTable(org.Hs.egSYMBOL)
m <- match(y$genes$EntrezGene, egSYMBOL$gene_id)
y$genes$Symbol <- egSYMBOL$symbol[m]
head(y$genes)
3.4) 过滤和归一化(Filtering and normalization)
过滤一:Different RefSeq transcripts for the same gene symbol count predominantly the same reads. So we keep one transcript for each gene symbol. We choose the transcript with highest overall count:
o <- order(rowSums(y$counts), decreasing=TRUE)
y <- y[o,]
d <- duplicated(y$genes$Symbol)
y <- y[!d,]
nrow(y)
过滤二:Normally we would also filter lowly expressed genes.For this data, all transcripts already have at least 50 reads for all samples of at least one of the tissues types.
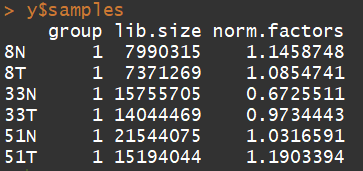
y$samples$lib.size <- colSums(y$counts) #Recompute the library sizes
###############################Use Entrez Gene IDs as row names:#####################
rownames(y$counts) <- rownames(y$genes) <- y$genes$EntrezGene
y$genes$EntrezGene <- NULL
归一化:TMM normalization is applied to this dataset to account for compositional difference between the libraries.
y <- calcNormFactors(y)
y$samples
3.5) 数据的探索(Data exploration)
样本间关系(samples for outliers and for other relationships)
plotMDS(y)
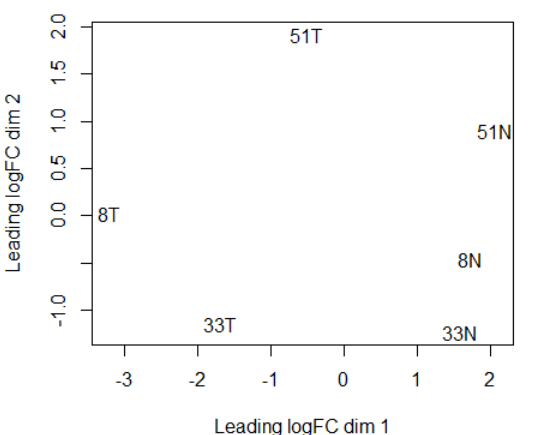
PC1将tumor和nomal组分开,PC2 大略和病号对应。也侧面体现了肿瘤组的异质性
--------------------------分组矩阵(根据实验设计、目的)--------------------------------
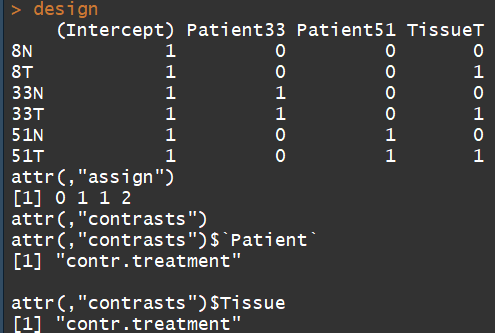
Here we want to test for differential expression between tumour and normal tissues within patients, i.e. adjusting for differences between patients.
Patient <- factor(c(8,8,33,33,51,51))
Tissue <- factor(c("N","T","N","T","N","T"))
data.frame(Sample=colnames(y),Patient,Tissue)
design <- model.matrix(~Patient+Tissue)
rownames(design) <- colnames(y)
design
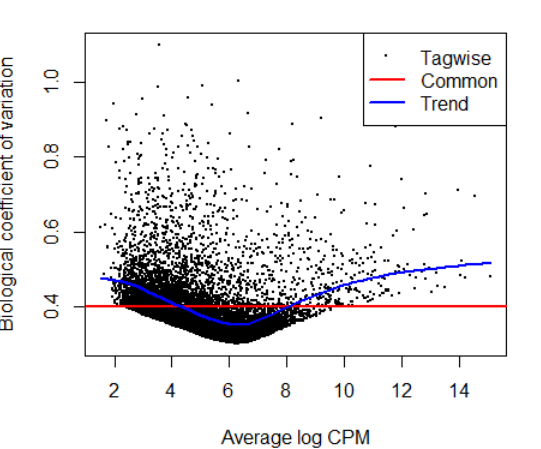
3.4)Estimating the dispersion(estimate the NB dispersion for the dataset.)
y <- estimateDisp(y, design, robust=TRUE)
y$common.dispersion #0.1594505
plotBCV(y)
-----------------------------------差异分析-----------------------------------------
3.5) 差异分析(Differential expression)
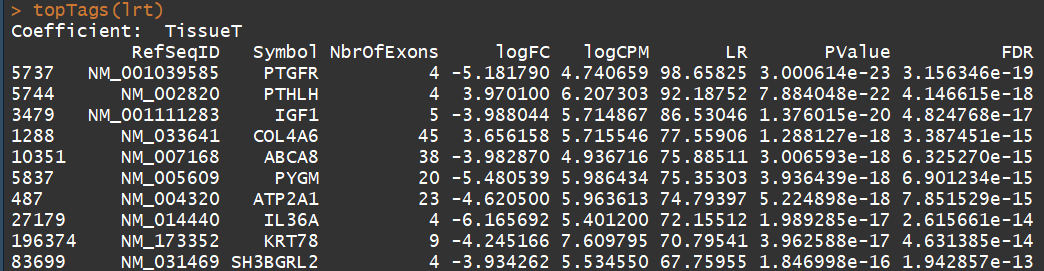
fit <- glmFit(y, design)
lrt <- glmLRT(fit)
topTags(lrt)
summary(decideTests(lrt))
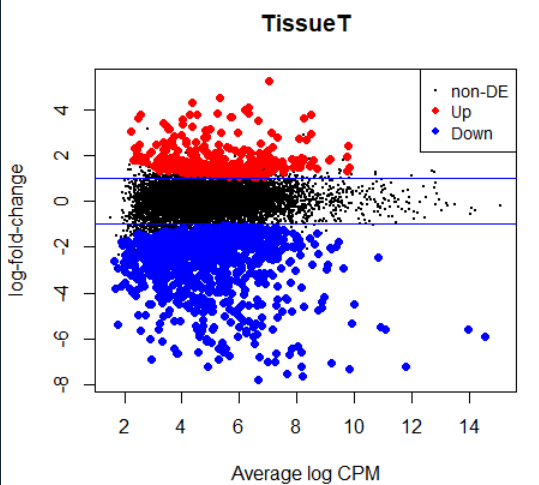
plotMD(lrt)
abline(h=c(-1, 1), col="blue")

------------------------------- Gene ontology analysis----------------------------------------
对上调的基因进行BP分析
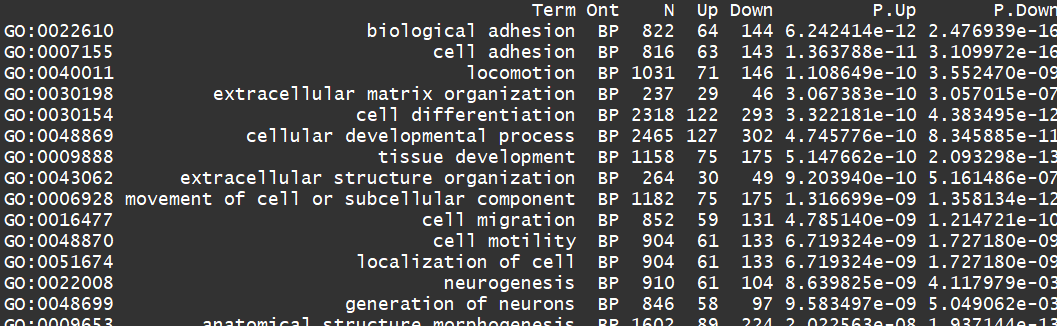
go <- goana(lrt)
topGO(go, ont="BP", sort="Up", n=30)
edgeR的更多相关文章
- cuffdiff 和 edgeR 对差异表达基因的描述
ASE又走到了关键的一步 要生成能决定是否有差异表达的table. 准备借鉴一下cuffdiff和edgeR 的结果 cuffdiff对差异表达基因的描述: 一共十四列: 第一列, test_id ...
- 简单使用DESeq2/EdgeR做差异分析
简单使用DESeq2/EdgeR做差异分析 Posted: 五月 07, 2017 Under: Transcriptomics By Kai no Comments DESeq2和EdgeR都 ...
- 使用limma、Glimma和edgeR,RNA-seq数据分析易如反掌
使用limma.Glimma和edgeR,RNA-seq数据分析易如反掌 Charity Law1, Monther Alhamdoosh2, Shian Su3, Xueyi Dong3, Luyi ...
- edgeR使用学习【转载】
转自:http://yangl.net/2016/09/27/edger_usage/ 1.Quick start 2. 利用edgeR分析RNA-seq鉴别差异表达基因: #加载软件包 librar ...
- 用TCGA收集的mRNA表达数据作差异表达
做差异表达的软件DEseq和edgeR所需要的数据格式必须是原始counts,经过normalization和log2后的数据都不适合,所以对于做差异表达计算的童鞋可以使用ExperimentHub下 ...
- sql是最成功的第四代语言
SQL发展的前世今生 很多年前,两名年轻的IBM研究员将一门关系型语言带到了数据库领域,旨在使用声明性的方式来操作数据.从Don Chamberlin和Ramond Boyce发表"SEQU ...
- RNA-seq标准化
你的 heatmap 可能用错数据了 (组间表达量标准化) http://www.genek.tv/article/24 RNA-seq的标准化方法罗列 https://www.jianshu.com ...
- 史上最全 | 39个RNAseq分析工具与对比
文献:Sahraeian S M E, Mohiyuddin M, Sebra R, et al. Gaining comprehensive biological insight into the ...
- RNA-seq中的基因表达量计算和表达差异分析
RNA-seq中的基因表达量计算和表达差异分析 差异分析的步骤:1)比对:2) read count计算:3) read count的归一化:4)差异表达分析: 背景知识:1)比对:普通比对: BWA ...
随机推荐
- Kibana 基础入门
原文地址:Kibana 基础入门 博客地址:http://www.extlight.com 一.前言 Kibana 是一个开源的分析和可视化平台,旨在与 Elasticsearch 合作.Kibana ...
- onload属性使用方法
onload事件属性是页面的图片文字等全部加载完毕后执行的事件 window.onload=fun1;function fun1(){ document.getElementsByTagName ...
- Bootstrap:百科
ylbtech-Bootstrap:百科 Bootstrap (Web框架) Bootstrap,来自 Twitter,是目前很受欢迎的前端框架.Bootstrap 是基于 HTML.CSS.Java ...
- 搭建GlusterFS文件系统
(1)环境准备 创建两个虚拟机配置如下 把仅主机第二张网卡配置如下: GlusterFS1 GlusterFS2 上传文件到opt目录下 文件内容如下 (2)GlusterFS安装配置 1.安装Glu ...
- Xss漏洞原理分析及简单的讲解
感觉百度百科 针对XSS的讲解,挺不错的,转载一下~ XSS攻击全称跨站脚本攻击,是为不和层叠样式表(Cascading Style Sheets, CSS)的缩写混淆,故将跨站脚本攻击缩写为XS ...
- Linux下apache2及模块mod_deflate等安装和配置
安装apache 1.wget http://archive.apache.org/dist/httpd/httpd-2.2.13.tar.gz 2.在安装目录 先让大家看看实际效果,请看下图10点中 ...
- API网关Kong系列(四)认证配置
目前根据业务需要先介绍2种认证插件:Key Authentication 及 HMAC-SHA1 认证 Key Authentication 向API添加密钥身份验证(也称为API密钥). 然后,消 ...
- javaScript中对象属性的访问
有两种方式访问对象属性,一个是点操作符(.),一种是中括号操作符([ ]). 当你知道属性的名称时,使用点操作符: var myObj = { prop1: "val1", pro ...
- js、C#获取当前url的参数值
之前很想做一些封装关于获取URL参数值方法,今天简单整理了一下js和后台代码获取url参数值的方法,有什么不好地方,还请大家包涵,代码如下: 1.JS处理URL参数值 <script langu ...
- 支持向量机(理论+opencv实现)
从基础开始讲起,没有这些东西看支持向量机真的很难! 1.拉格朗日乘子(Lagrangemultiplier) 假设需要求极值的目标函数(objectivefunction)为f(x,y),限制 ...