circRNA
一、circRNA序列提取
环状RNA (circRNA)是一类不具有 5' 末端帽子和 3' 末端 poly(A)尾巴、并以共价键形成环形结构的非编码 RNA 分子。 环状RNA (circRNA) 是区别于传统线性 RNA 的一类新型 RNA,大量存在于真核转录组中且表达具有时空特异性。在调控基因转录、作为疾病诊断marker等方面具有重要的研究和临床意义。
在预测circRNA时,都是检测breakpoint 处的reads 数,最后给出的环状RNA的ID 都是诸如 chr14:106994222-107183708 这样给出了起始和终止位置;对于某一个基因来说,其可能产生的circRNA的类型是多样的,以下图为例进行说明

1)由单个外显子组成的环状RNA, 比如
2)有多个外显子组成的环状RNA, 比如

以上的两种circRNA在序列提取时都非常容易,只需要将circRNA的起始和终止位置能够和某些外显子正好对应上,那么就可以确定其序列就是起始外显子和终止外显子之间的所有外显子构成的序列
3)只由内含子组成的环状RNA

这种环状RNA也可以方便的提取序列,直接确定起始和终止位置在基因组上的位置,将对应的序列提取出来即可
4)起始外显子和终止外显子之间有多个外显子,比如
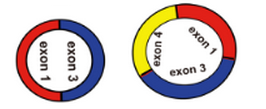
5)起始外显子和终止外显子之间有内含子,比如
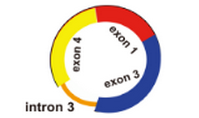
预测环状RNA时,只能够确定起始外显子和终止外显子,却不能确定在该circRNA中间到底有哪几个外显子,而且到底包不包含内含子序列,由于可变剪切的存在,可能存在多个外显子,也可能包含内含子,是不能够准确的提取circRNA对应的序列;能够做的只是将包括起始外显子和终止外显子以及之间的所有外显子连起来作为circRNA的序列
以上面的exon1-exon4 之间形成的环状RNA为例,我们只能将exon1-exon2-exon3-exon4的序列作为该环状RNA的序列,但是和实际的环状RNA的序列肯定是存在误差的;
目前分析手段没办法很好的解决这个问题,也许随着对环状RNA认识的加深和分析方法的改进,可以准确的识别circRNA的序列;为了准确的确定circRNA的序列,只能是针对breakpoint 两边的序列设计特异性引物,将circRNA 扩增出来,再测序,准确的识别序列;
二、提取fa
假设存在一个exon的bed文件,以及参考基因组的fa序列和gtf文件,如何根据reference和gtf文件,提取这个exon_bed文件中区间内的所有外显子序列。如exon_bed文件中有一个区间1 16366079 16509408 1:16366079|16509408 exon,那么如何将1:16366079-16509408内所有外显子的序列提取出来,然后进行拼接。先利用bedtools intersect对exon_bed和gtf文件求一个交集(注意:exon_bed和gtf文件中获取的所有exon区间最好先进行排序),以获得该区间内的所有外显子,如下所示:
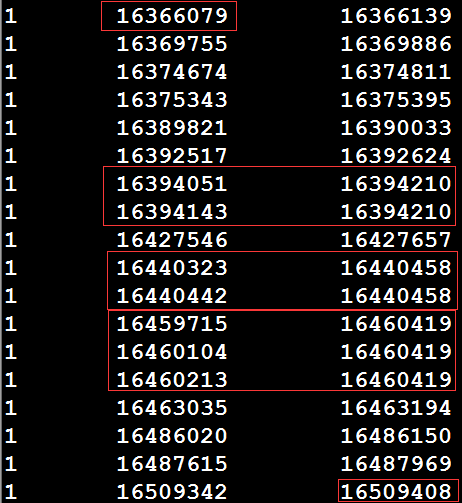
从上图可以看出,由于可变剪切的存在,导致存在多个转录本,从而exon区间存在over_lap的情况,上图需要红框内的行需要进行预处理,选择最大区间。得到下图序列:
获取最大区间,即bed文件进行处理后,再利用bedtools getfasta从参考序列中提取该区间内的序列,然后进行合并,产生1:16366079-16509408之间所有外显子序列。其中脚本处理如下,使用方法:bash process.sh path_of_refgenome
#!/bin/bash
genome_path=$1
bed_over_path='.' #从Mus_musculus.GRCm38.84.gtf文件中获取所有exon区间,并排序去重
grep -v '^#' $genome_path/Mus_musculus.GRCm38.84.gtf | grep -w 'exon' | cut -f1,4-5 | sort -V | uniq > all_exon_from_gtf if [ ! -d temp ]
then
mkdir temp
fi >temp/exon_all.fa
>exon_all.fa #step1:读取exon_bed的每一行,每一行相当于一个circRNA,将每一行值付给相应变量
#step2:生成过程文件:chrom_start_end_bed记录了exon_bed的每一行信息
#step3:bedtools intersect获取all_exon_from_gtf文件和chrom_start_end_bed交集
#step4:bed_over.py对step3产生的交集chrom_start_end_over_lap.bed文件中的overlap行选取最大外显子区间
#step5:使用bedtools getfasta获取chrom_start_end_over_lap_bed文件对应的fa序列,即完成对exon_bed文件一行的序列提取工作
#step6:进行合并序列操作,先将exon_bed中的>header写入chrom_start_end.fa
#step7:将bedtools getfasta获取的exon序列去掉>行后再去掉换行符,并追加到step6对应的chrom_start_end.fa中
#setp8:最后合并exon_bed文件每一行对应的fa序列,生成exon_all.fa序列文件
while read chrom start end start_end type
do
echo -e "$chrom\t$start\t$end" > "$chrom"_"$start"_"$end"_bed
bedtools intersect -a $genome_path/all_exon_from_gtf -b "$chrom"_"$start"_"$end"_bed > "$chrom"_"$start"_"$end"_over_lap.bed python $bed_over_path/bed_over.py "$chrom"_"$start"_"$end"_over_lap.bed > "$chrom"_"$start"_"$end"_over_lap_bed
bedtools getfasta -fi $genome_path/Mus_musculus.GRCm38.dna.primary_assembly.fa -bed "$chrom"_"$start"_"$end"_over_lap_bed -fo "$chrom"_"$start"_"$end".fa echo ">$start_end" > temp/"$chrom"_"$start"_"$end".fa
grep -v '>' "$chrom"_"$start"_"$end".fa | tr -d "\n" >> temp/"$chrom"_"$start"_"$end".fa
echo '' >> temp/"$chrom"_"$start"_"$end".fa cat temp/"$chrom"_"$start"_"$end".fa >> exon_all.fa
rm -f "$chrom"_"$start"_"$end"_bed
rm -f "$chrom"_"$start"_"$end"_over_lap.bed
rm -f "$chrom"_"$start"_"$end"_over_lap_bed
rm -f "$chrom"_"$start"_"$end".fa
done< exon_bed
其中的bed_over.py脚本是用来处理over_lap行的,脚本见下面:
#!/bin/python
#coding=utf-8
import sys
pre_start = 0
pre_end = 0
flag = 1
pre_chrom = ''
f_open = open(sys.argv[1]) #逐行处理待处理的存在over_lap行的bed文件
#采取的方法是在读取下一行,进行over_lap判断
#需要先保留上一行的chrom,start,end
for each in f_open:
array = each.strip().split('\t')
# chrom = array[3].split(':')[0]
# start = int(array[3].split(':')[1].split('|')[0])
# end = int(array[3].split(':')[1].split('|')[1])
chrom = array[0]
start = array[1]
end = array[2]
if 1 == flag:
pre_start = start
pre_end = end
pre_chrom = chrom
flag = flag + 1
continue
else:
if chrom == pre_chrom:#下一行chrom与上一行相同,才有必要比较是否存在over_lap
if start > pre_end:#下一行start比上一行的end还大,就不存在over_lap,需要将上一行写入文件
#print str(pre_chrom)+'\t'+str(pre_start)+'\t'+str(pre_end)
print '\t'.join([str(pre_chrom),str(pre_start),str(pre_end)])
pre_start = start
pre_end = end
pre_chrom = chrom
flag = flag + 1
continue
elif start <= pre_end:#下一行start比上一行的end小,所以存在over_lap情况,更新end为该行end,替换上一行的end
pre_end = end
pre_chrom = chrom
flag = flag + 1
continue
else:#下一行chrom与上一行chrom不同,直接进行上行写入操作
#print str(pre_chrom)+'\t'+str(pre_start)+'\t'+str(pre_end)
print '\t'.join([str(pre_chrom),str(pre_start),str(pre_end)])
pre_start = start
pre_end = end
pre_chrom = chrom
flag = flag + 1
continue f_open.close()
#最后一行由于没有下一行了,直接进行写入操作
#print str(pre_chrom)+'\t'+str(pre_start)+'\t'+str(pre_end)
print '\t'.join([str(pre_chrom),str(pre_start),str(pre_end)])
circRNA的更多相关文章
- circRNA 中的ALU 重复元件
circRNA 最初研究的很少,只有很小一部分基因有检测到circRNA, 当时都认为是剪切错误形成的,对于其功能也没人去研究:学者对人类的成纤维细胞进行转录组测序,构建去核糖体文库, 同时采用了RN ...
- circRNA 在人和小鼠脑组织中的表达
circRNA 是一类动物体内的内源性的RNA,尽管circRNA的种类丰富,但是其在神经系统中的 功能,并不清楚.科学家通过对人和小鼠的不同脑部组织的RNA 测序,发现了上千种circRNA,经过分 ...
- circRNA研究手册
环状RNA(circRNA)研究技术手册.doc.pdf (转自:汉恒生物)
- 利用circpedia 数据库探究circRNA的可变剪切
circpedia 中收录了利用circexplorer 软件识别到的circRNA, 覆盖了人,小鼠,鸟类,昆虫多个物种的多种细胞系的数据 官网链接如下: http://www.picb.ac.cn ...
- find_circ 识别circRNA 的原理
find_circ 通过识别junction reads 来预测circRNA 和参考基因组比对完之后,首先剔除和基因组完全比对的reads,保留没比对上的reads, 这部分reads 直接比是比对 ...
- CIRI 识别circRNA的原理
CIRI 根据circRNA 连接点处的reads来识别circRNA, 在连接点处的reads 其比对情况非常特殊: CIRI 根据3种模型来识别circRNA, 连接点处的read 叫做junct ...
- circRNA 序列提取中的难点
在预测circRNA时,都是检测breakpoint 处的reads 数,最后给出的环状RNA的ID 都是诸如 chr14:106994222-107183708 这样的形式,给出了起始和终止位置: ...
- CircRNA 环化RNA
2016国自然新秀CircRNA的研究策略和分析
- circRNA数据库的建立
circRNA数据库的建立 wget http://circbase.org/download/human_hg19_circRNAs_putative_spliced_sequence.fa.g ...
随机推荐
- Linux环境C程序设计
Linux基础 常用shell命令 命令 说明 命令 说明 man 查看联机帮助 ls 查看目录及文件列表 cp 复制目录或文件 mv 移动目录或文件 cd 改变文件或目录 rm 删除文件或目录 mk ...
- PAT 1076 Wifi密码
https://pintia.cn/problem-sets/994805260223102976/problems/994805262622244864 下面是微博上流传的一张照片:“各位亲爱的同学 ...
- Node buffer模块缓冲区
//1:字节 byte//1024b = 1kb [千字节]//1024kb = 1mb [兆字节]//1024mb = 1gb [吉字节]//1024gb = 1tb [梯]//1024tb = 1 ...
- Delphi中封装ADO之我重学习记录
delphi adodataset ctstatic 数据是缓存在服务器端还是客户端 答:客户端,开启本地缓存功能后,就能数据在本地批量修改后,再批量提交,减少了网络传送 原创,专业,图文 Del ...
- 使用nmon进行系统监控
一.下载并安装: 下载地址:http://nmon.sourceforge.net/pmwiki.php?n=Site.Download 下载版本:nmon16g_x86.tar.gz 不用的Li ...
- python之文件目录操作
代码示例: # 改变当前目录操作 import os cur = os.curdir print("1.当前目录相对路径:", cur) par = os.pardir print ...
- asp.net 后台<%@ Page%> page指令属性
aspx文件有如下一行代码: <%@ Page Language="C#" AutoEventWireup="true" CodeBehind=" ...
- BZOJ1087[SCOI2005]互不侵犯——状压DP
题目描述 在N×N的棋盘里面放K个国王,使他们互不攻击,共有多少种摆放方案.国王能攻击到它上下左右,以及左上左下右上右下八个方向上附近的各一个格子,共8个格子. 输入 只有一行,包含两个数N,K ( ...
- 经典Java面试题收集(二)
经典的Java面试题(第二部分),这部分主要是与Java Web和Web Service相关的面试题. 96.阐述Servlet和CGI的区别? 答:Servlet与CGI的区别在于Servlet处于 ...
- 2.9 C++使用默认参数的构造函数
总结: 默认参数的构造函数,其默认参数必须置于参数列表的结尾. 设计类的构造函数的时候最好不要同时是用构造函数的重载和带参数的构造函数. 我们可以想象一个这样的场景:某一天书店整理库存,发现了一些非常 ...