链终止法|边合成边测序|Bowtie|TopHat|Cufflinks|RPKM|FASTX-Toolkit|fastaQC|基因芯片|桥式扩增|
生物信息学
Sanger采用链终止法进行测序
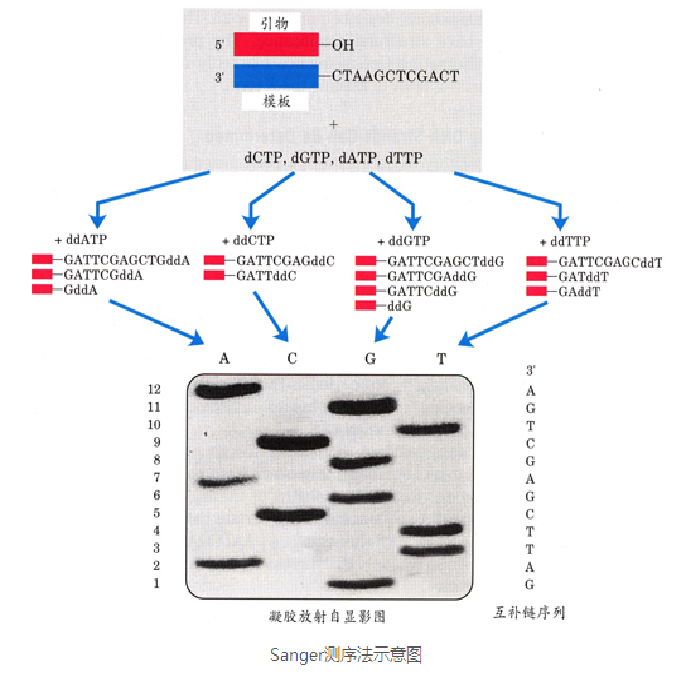
带有荧光基团的ddXTP+其他四种普通的脱氧核苷酸放入同一个培养皿中,例如带有荧光基团的ddATP+普通的脱氧核苷酸A、T、C、G放入同一个培养皿,以此类推,存在4种不同类型碱基的识别机制,同时,该ddXTP一旦结合在互补链上则会迫使复制停止。
高通量测序是二代测序,先建库后测序:
建库方法:
单末端测序:将DNA双链打碎并接上接头序列,通过改变条件使双链变单链,将待测的单链固定在flowcell上,再加入游离的脱氧核苷酸,采用边合成边测序方法比配并测得互补链,最后冲走互补链。
双末端测序:将DNA双链打碎并接上接头序列,通过改变条件使双链变单链,将待测的单链固定在flowcell上,采用桥式扩增(即将模板链和互补链单链都成簇,用酶切,就只剩下模板链和两端引物),通过桥式扩增加强信号。
它的碱基识别是在链终止法的基础上,采用边合成边测序方法:
带有荧光基团的ddXTP+其他三种普通的脱氧核苷酸放入同一个培养皿中,例如带有荧光基团的ddATP+普通的脱氧核苷酸T、C、G放入同一个培养皿,以此类推,存在4种不同类型碱基的识别机制,同时,该ddXTP一旦结合在互补链上则会迫使复制停止。 模板链通过碱基互补配对原则得到互补链,该互补链停止于需识别碱基上,通过测试最后一位碱基所携带的荧光基团,从而确定模板链对应的碱基种类。在确认了该位置的碱基种类之后,会加羟基去去除荧光基团,并使得链又不断复制下去,通过识别后的照片来得到最终的序列组成。
高通量测序的特点是数据量大。
Fasta:
>序列名字
序列本身
Fastq:
@序列信息
序列本身
@序列信息
序列质量:由碱基错误率算得,每个碱基对应一个质量,将具体数值用ASCII表示出来
质量指标:Q20、Q30、Q40
基因芯片和高通量测序的比较:
基因芯片原理:
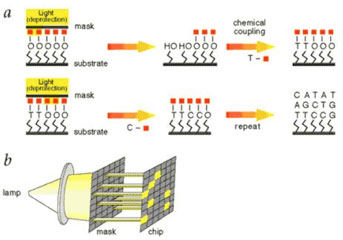
通过探针捕获游离的带有荧光基团的脱氧核苷酸,然后使其荧光标记发出荧光,辨别荧光强度确定碱基种类并加入羟基使其不再发光,再以此类推确定另外一种荧光种类。
二代测序:双末端测序;边合成边测序
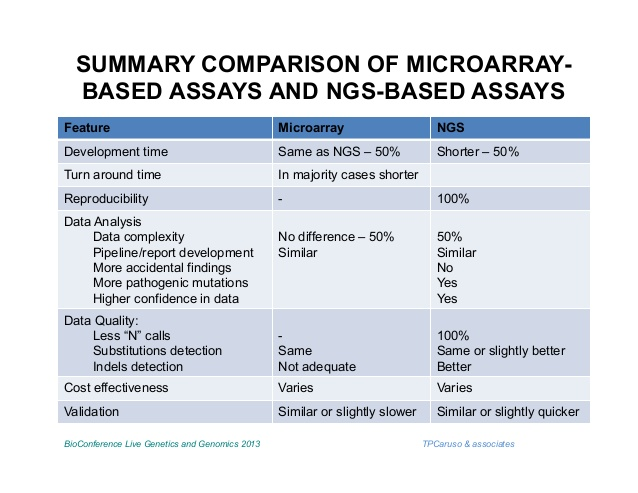
区别:前期样本制备
基因表达:相当
数据分析流程:
图片数据(识别荧光基团测序)----序列信息(fastq)------质控-----assembly----analysis----annotation
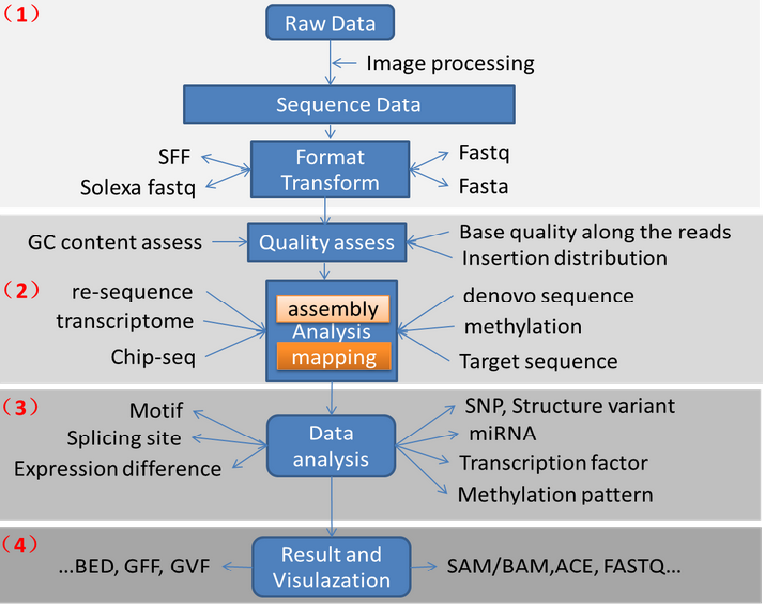
Raw data------质控fastaQC------去adoptor/linker(引物)------使用barcode排序------FASTX-Toolkit质控(某特殊位置之后的;低于平均值的)
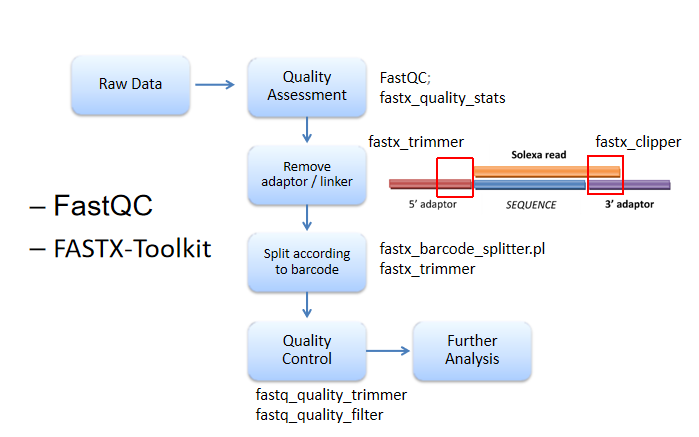
fastaQC:FASTQC checks whether a set of sequence reads in a .fastq fifile exhibit any unusual qualities.

FASTX-Toolkit:In this section of the tutorial we will be using FASTX-Toolkit (http://hannonlab.cshl.edu/fastx_toolkit/) to fifilter and trim sequences based on quality.
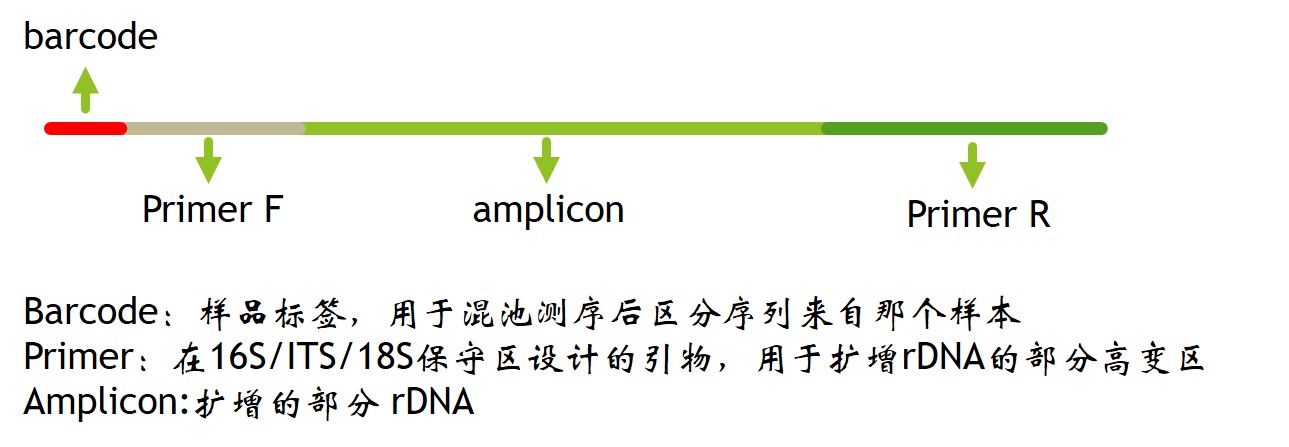
调研(低深度测序)主要用于找到合理建库方法----框架(高深度测序)----精细测序,进行修补
应用:
个体重测序----精准医疗----突变
重测序数据分析:GATK:人群队列测----发现SNP并汇总----发现群体高发SNP
转录组测序:
1.Small RNA seq表达
2.RNA seq表达-:--->Bowtie(短片段RNA比genome)---->Tophat(找到splice junction)----->cufflinks(找到可变剪接)
Bowtie is an ultrafast, memory-efficient short read aligner geared toward quickly aligning large sets of short DNA sequences (reads) to large genomes.
TopHat is a fast splice junction mapper for RNA-Seq reads.
Cufflinks assembles transcripts, estimates their abundances, and tests for differential expression and regulation in RNA-Seq samples.
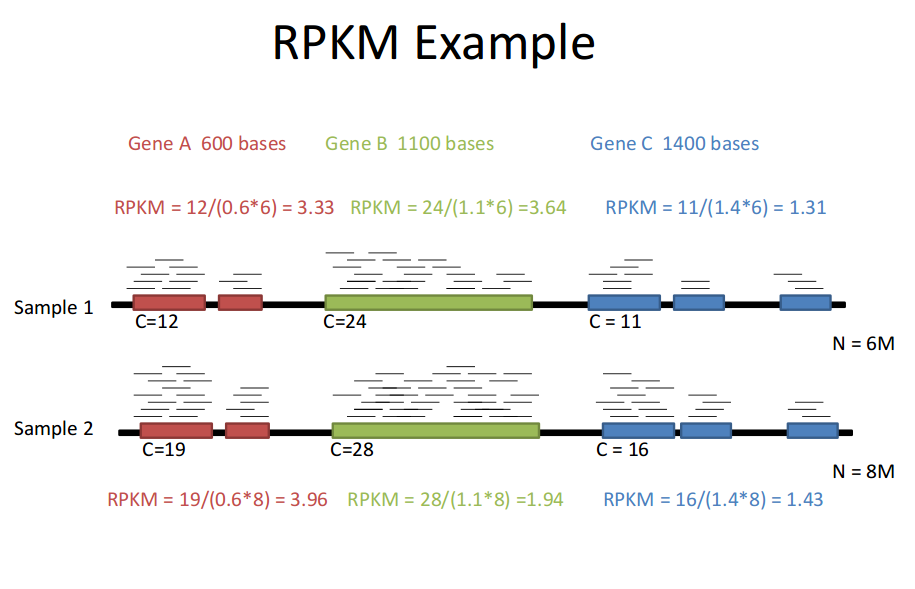
RPKM (Reads Per Kilobase of transcript per Million mapped reads)
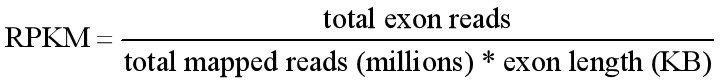
Total Exon reads:比到某个sample上的read数;
total mapped reads:比到某个sample上某gene exon的reads数
exon length:某gene exon长度
所以,
sample之间同一gene比较的是(gene mapped reads数/sample mapped reads数)
同一sample 的不同gene之间比较的是(gene mapped reads数/gene oxen length)
Chip-seq:发现转录因子的结合位点
链终止法|边合成边测序|Bowtie|TopHat|Cufflinks|RPKM|FASTX-Toolkit|fastaQC|基因芯片|桥式扩增|的更多相关文章
- 使用Tophat+cufflinks分析差异表达
使用Tophat+cufflinks分析差异表达 2017-06-15 19:09:43 522 0 0 使用TopHat+Cufflinks的流程图 序列的比对是RNA分析 ...
- SAGE|DNA微阵列|RNA-seq|lncRNA|scripture|tophat|cufflinks|NONCODE|MA|LOWESS|qualitile归一化|permutation test|SAM|FDR|The Bonferroni|Tukey's|BH|FWER|Holm's step-down|q-value|
生物信息学-基因表达分析 为了丰富中心法则,研究人员使用不断更新的技术研究lncRNA的方方面面,其中技术主要是生物学上的微阵列芯片技术和表达数据分析方法,方方面面是指lncRNA的位置特征. Bac ...
- tophat cufflinks cuffcompare cuffmerge 的使用
Cole Trapnell said: there are three strategies: 1) merge bams and assemble in a single run of Cuffli ...
- illumina SBS测序详解
illumina SBS测序详解 2018年01月02日 09:33:56 sixu_9days 阅读数:9789 标签: 生物信息学二代测序 更多 个人分类: 测序原理 最近回头重新看了illl ...
- Next generation sequencing (NGS)二代测序数据预处理与分析
二代测序原理: 1.DNA待测文库构建. 超声波把DNA打断成小片段,一般200--500bp,两端加上不同的接头2.Flowcell.一个flowcell,8个channel,很多接头3.桥式PCR ...
- 解读生命密码的基本手段 ——DNA测序技术的前世今生
解读生命密码的基本手段 ——DNA测序技术的前世今生 任鲁风 于军 (中国科学院基因组科学及信息重点实验室,北京基因组研究所) DNA(脱氧核糖核酸)和RNA(核糖核酸)是生命体的两种最基本组成物质 ...
- 第三代PacBio测序技术的测序原理和读长
针对PacBio单分子测序——第三代测序技术的测序原理和读长 DNA基因测序技术从上世纪70年代起,历经三代技术后,目前已发展成为一项相对成熟的生物产业.测序技术的应用也扩展到了生物.医学.制 ...
- TopHat
What is TopHat? TopHat is a program that aligns RNA-Seq reads to a genome in order to identify exon- ...
- 单细胞测序技术(single cell sequencing)
单细胞测序技术(single cell sequencing) 2018-03-02 11:02 来源: 一呼百诺 点击次数:6587关键词: 前言 单细胞生物学最近几年是非常热门的研究方向 ...
随机推荐
- [浅学]POST、GET、PUT、DELETE 请求
HTTP定义了与服务器交互的不同的方法,最基本的是POST.GET.PUT.DELETE,与其比不可少的URL的全称是资源描述符,我们可以这样理解: url描述了一个网络上资源,而post.get.p ...
- 17。3.12---re模块--正则表达式操作指南
1----python re模块(Regular Expressioin正则表达式)提供了一个与perl等编程语言类似的正则匹配操作,他是一个处理python字符串的强有力的工具,有自己的语法和独立的 ...
- 吴裕雄--天生自然 PHP开发学习:表单 - 验证邮件和URL
$name = test_input($_POST["name"]); if (!preg_match("/^[a-zA-Z ]*$/",$name)) { $ ...
- C - Monitor CodeForces - 846D (二维前缀和 + 二分)
Recently Luba bought a monitor. Monitor is a rectangular matrix of size n × m. But then she started ...
- UML-领域模型-如何找到概念类
有3个方法 方法1:对已有的重用和修改(这是最好的方法) 重用和修改现有模型.这些模型来源于之前的项目.网上的 方法2:使用分类列表 从网上搜索该领域的常见分类,或参考书籍Martin Fowler的 ...
- SOA架构设计分析
SOA(Service-Oriented Architecture,面向服务的架构)是一个组件模型,它将应用程序的不同功能单元(称为服务)进行拆分,并通过这些服务之间定义良好的接口和契约联系起来. S ...
- Linux 安装python3.x步骤
本文转发自博客园非真的文章,内容略有改动 本文已收录至博客专栏linux安装各种软件及配置环境教程中 linux系统本身默认安装有2.x版本的python,版本x根据不同版本系统有所不同,通过pyth ...
- vue 中使用print.js 打印遇到的问题 ?
不管怎么设置打印部分的 margin和height 仍会在预览时多出一张空白页?求各位大佬遇到过的请留言谢谢!
- visual studio2019下动态链接库的制作
打开visual studio2019创建动态链接库项目,项目名称为20199324dll 然后定义宏:在头文件中定义即可,宏的作用的是允许该函数能够被外部访问,并直接调用.代码如下: // pch. ...
- linux查看显卡
查看 nvidia 显卡 $ lspci | grep -i nvidia 02:00.0 3D controller: nVidia Corporation Device 1023 (rev a1) ...