fasta/fastq格式解读
1)知识简介
--------------------------------------------------------
1.1)测序质量值
首先在了解fastq,fasta之前,了解一下什么是质量值。phred软件在对reads进行base calling的时候会给出每一个碱基的质量值,这个质量值的计算与测序预期错误率相关(estimated probability of error):
Phred Quality Score Probability of incorrect base call Base call accuracy
10 1 in 10 90 %
20 1 in 100 99 %
30 1 in 1000 99.9 %
40 1 in 10000 99.99 %
50 1 in 100000 99.999 %
除此之外还有solexa标准,即将p换成了p/(1-p),其他完全按照sanger的定义来做。当测序质量很高的情况下两种形式几乎没区别,但低质量的碱基则有区别了(见下图)

Qscore与p之间的关系,其中红线表示Q=-10 log10p标准,黑色实线表示Q=-10 log10p/(1-p)标准。
1.2)ACII码
为了方便储存及可读这些信息,利用可打印的ACII码将这些质量值转化为单字符single characters (or bytes)。ASCII 字符集,最基本的包含了128 个字符。其中前 32 个, 0-31 ,即 0x00-0x1F ,都是不可见字符,这些字符,为控制字符。可见字符为32–126。sanger-fastaq格式用 ASCII 33–126 来表示phred 质量值 0 到93 。举例来说:一般地,碱基质量从0-40,既ASCii码为从 “!”(0+33)到“I”(40+33)。如果某碱基测序出错的概率为0.001,则Q应该为30。则30+33=63,那么63对应的ASCii码为“?”,在第四行中该碱基对应的质量代表值即为“?”。

2)fastq格式
fastq格式是一个文本格式用于贮存生物学序列及其相应质量值(通常是核酸序列的)。为了简介,这些序列以及质量信息使用ASCII字符标示。该格式最初由Sanger开发,目的是将FASTA序列与质量数据放到一起,目前已经成为高通量测序结果的事实标准。通常fastq文件中每一个序列含有4行信息(如下):
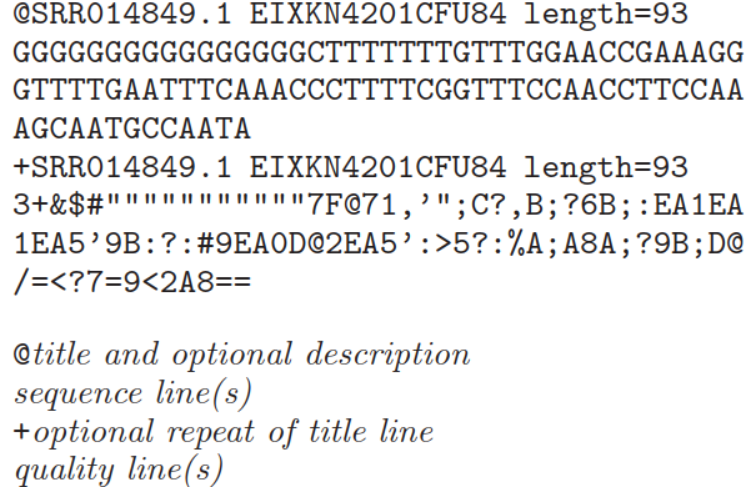
第一行:序列标识,以‘@’开头。格式比较自由,允许添加注释等相关的描述信息,描述信息以空格分开。如示图中描述信息加入了NCBI的另一个ID名称,及长度信息
第二行:表示序列信息,制表符或者空格不允许出现。一般是明确的DNA或者RNA字符,通常是大写,在一些文本文件中,小写或者大小写混杂或者含有gap符号是有特殊含义。
第三行:用于将测序序列和质量值内容分离开来。以‘+’开头,后面是描述信息等,或者什么也不加。如果“+”后面有内容,该内容与第一行“@”后的内容相同;
第四行:表示质量值,每个字符与第二行的碱基一一对应,按照一定规则转换为碱基质量得分,进而反映该碱基的错误率,因此字符数必须和第二行保持一致。对于每个碱基的质量编码标示,不同的软件采用不同的方案 。目前有5种:
1、Sanger,Phred quality score:值的范围从0到93,对应的ASCII码从33到126,但是对于测序数据(raw read data)质量得分通常小于60,序列拼接或者mapping可能用到更大的分数。
2、Solexa/Illumina 1.0, Solexa/Illumina quality score:值的范围从-5到62,对应的ASCII码从59到126,对于测序数据,得分一般在-5到40之间;
3、Illumina 1.3+,Phred quality score:值的范围从0到62对应的ASCII码从64到126,低于测序数据,得分在0到40之间;
4、Illumina 1.5+,Phred quality score:但是0到2作为另外的标示,详见http://solexaqa.sourceforge.net/questions.htm#illumina
5、Illumina 1.8+
不同的标准之间可以相互转化换,感兴趣可以自己查资料,这里不做详细介绍。注意:第二行@字符,第三行+字符,在第四行质量值中会出现,有时也会在行首出现,因此在处理fastq格式的时候要格外的关注。
3)fasta格式
------------------------------------
3.1)fasta格式最初来自FASTA软件包,也是一种文本格式,以单字符( single-letter codes)贮存核酸或者蛋白序列信息,允许在序列前加注释信息。由2部分信息组成:
>gi|5524211|gb|AAD44166.1| cytochrome b [Elephas maximus maximus]
LCLYTHIGRNIYYGSYLYSETWNTGIMLLLITMATAFMGYVLPWGQMSFWGATVITNLFSAIPYIGTNLVEWIWGGFSVDKATLNRFFAFHFILPFTMVALAGV
HLTFLHETGSNNPLGLTSDSDKIPFHPYYTIKDFLGLLILILLLLLLALLSPDMLGDPDNHMPADPLNTPLHIKPEWYFLFAYAILRSVPNKLGGVLALFLSIV
IGLMPFLHTSKHRSMMLRPLSQALFWTLTMDLLTLTWIGSQPVEYPYTIIGQMASILYFSIILAFLPIAGXIENY
第一部分:以>号开始,紧接着序列的标识符 ,注意区分大小写,且不能出现空格,空格表示序列标识符结束; 随后是序列的描述信息。
第二部分:以序列本身信息,使用既定的核苷酸或氨基酸编码符号,大小写都可以。直到遇到下一个>结束。所有来源于NCBI的序列都有一个gi号“gi|gi_identifier”,gi号由数字组成,具有唯一性。一条核酸或者蛋白质改变了,将赋予一个新的gi号(这时序列的接收号可能不变)。gi号后面是序列的标识符,标识符由序列来源标识、序列标识(如接收号、名称等)等几部分组成,他们之间用“|”隔开,如果某项缺失,可以留空但是“|”不能省略。
3.2)fasta格式在拓展的文件命名中,一般会约定俗成,具体见下表格:
| Extension | Meaning | Notes |
|---|---|---|
| fasta | generic fasta | Any generic fasta file. Other extensions can be fas, fa, seq, fsa |
| fna | fasta nucleic acid | Used generically to specify nucleic acids. |
| ffn | FASTA nucleotide of gene regions | Contains coding regions for a genome. |
| faa | fasta amino acid | Contains amino acids. A multiple protein fasta file can have the more specific extension mpfa. |
| frn | FASTA non-coding RNA | Contains non-coding RNA regions for a genome, in DNA alphabet e.g. tRNA, rRNA |
4)习题(fq练习文件已boweti2中的示例文件reads_1.fq)
--------------------------------------------------------------------------------------------------
4.1) fq文件中的质量值是如何产生的?
4.2) fq的质量在转化成ACII码的时候,为什么不选择前32个?(0-32)
4.3) fq的质量值在转化成ACII码的时候,为什么不从32开始,而是从33开始?
4.4)在统计fq有多少条序列的时候能不能直接grep '@' read_1.fq | wc -l ?为什么?
4.5)在sanger和solexa标准中,测序的错误率与质量值之间的差别在哪里?
4.6)fasta格式起源于什么地方?4.7)fasta文件的命名有没有特殊的含义?
4.8)fasta序列标识符是如何对应来自不同的数据库来源的?
4.9)转换fasta与fasta的软件有哪些?
4.10)fasta中的字母大小写有没有特殊的含义?
4.11) 统计reads_1.fq文件中共有多少条序列信息
4.12)输出所有的reads_1.fq文件中的标识符(即以@开头的那一行)
4.13)计算reads_1.fq 所有的reads中N的总数
4.14)统计reads_1.fq 中测序碱基为Q30的含量
4.15)统计reads_1.fq 中测序碱基质量为所有大于Q20的碱基含量
4.16)将reads_1.fq转为reads_1.fa文件(即将fastq转化为fasta)
4.17)计算reads_1.fa文件中GC数量
4.18)统计文件中reads_1.fa碱基总数
4.19)计算reads_1.fa文件中GC含量百分比
4.20)过滤掉reads_1.fa文件中N含量超过10%的reads,并统计有多少条
5) 参考资源
--------------------------------------------------------------
The Sanger FASTQ file format for sequences with quality scores, and the Solexa/Illumina FASTQ variants
https://en.wikipedia.org/wiki/FASTQ_format
https://en.wikipedia.org/wiki/FASTA
http://boyun.sh.cn/bio/?p=1901
https://blog.csdn.net/open2open2/article/details/26706969
fasta/fastq格式解读的更多相关文章
- 文件格式——fastq格式
fastQ格式 FASTQ是一种存储了生物序列(通常是核酸序列)以及相应的质量评价的文本格式. 他们都是以ASCII编码的.现在几乎是高通量测序的标准格式.NCBI Short Read Archiv ...
- FASTQ格式
FASQT格式是用于存储生物序列(通常是核苷酸序列)及其相应的碱基质量分数的一种文本格式.为简洁起见,序列字母和质量分数均使用单个ASCII字符进行编码.最初由Wellcome Trust Sange ...
- mismatch位置(MD tag)- sam/bam格式解读进阶
这算是第二讲了,前面一讲是:Edit Distance编辑距离(NM tag)- sam/bam格式解读进阶 MD是mismatch位置的字符串的表示形式,貌似在call SNP和indel的时候会用 ...
- 非IMU模式下DML语句产生的REDO日志内容格式解读
实验内容:非IMU模式下DML语句产生的REDO日志内容格式解读 最详细的解读是UPDATE的. 实验环境准备 11G中默认是开启IMU特性的,做此实验需要关闭此特性. alter system se ...
- 利用kseq.h parse fasta/fastq 文件
在分析中经常需要统计fasta/fastq文件的序列数和碱基数, 但是没有找到一些专门做这件事的小工具,可能是这个功能太简单了: 之前用自己写的perl的脚本统计这些信息, 当fastq文件非常大时, ...
- Spark直接读入fastq格式的数据
输入文件: fastq格式 输出结果: kmer的频数和对应的kmer类型 系统环境Ubuntu单机版17.01 spark版本2.7 此次测试主要用到了RDD的函数foreach和zipWithIn ...
- 创世区块配置文件genesis.json的格式解读
创世区块配置文件genesis.json的格式解读 中文网站上关于genesis 的解析大多数都来自于这个Gist:Ethereum private network configuration gui ...
- fastx tookit 操作fasta/fastq 文件 (1)
准备测试文件 test.fq, 包含4条fastq 文件,碱基编码格式为phred64; @FC12044_91407_8_200_406_24 NTTAGCTCCCACCTTAAGATGTTTA + ...
- 将fasta fastq文件线性化处理
将fasta文件线性化处理 awk '/^>/ {printf("%s%s\t",(N>0?"\n":""),$0);N++;n ...
随机推荐
- <dedecms>织梦内页调用会员信息
1.织梦CMS v5.7调用文章所属会员信息标签 打开官方默认模板article_artcile.htm,我们可以提取出如下代码: {dede:memberinfos} 会员头像:<a h ...
- linux 异步信号的同步处理方式
关于代码的可重入性,设计开发人员一般只考虑到线程安全,异步信号处理函数的安全却往往被忽略.本文首先介绍如何编写安全的异步信号处理函数:然后举例说明在多线程应用中如何构建模型让异步信号在指定的线程中以同 ...
- unittest框架模版 (含智能执行类下面所有用例并出报告)
基础框架一: import unittest class denglu(unittest.TestCase): def setUp(self): #每次执行测试用例前操作步骤 self.verific ...
- 垃圾收集器之:CMS收集器
HotSpot JVM的并发标记清理收集器(CMS收集器)的主要目标就是:低应用停顿时间.该目标对于大多数交互式应用很重要,比如web应用.在我们看一下有关JVM的参数之前,让我们简要回顾CMS收集器 ...
- position和margin的绝对定位和相对定位
电脑桌面清理干净之后,果然快了很多,桌面上的东西会占用内存,导致电脑变慢,以前我看到表姐的电脑桌面堆满了东西,我就在心里默默的鄙视不懂玩电脑的人,现在我竟然也养成了这种坏毛病..保存东西的时候放在桌面 ...
- 利用ubuntu的alias命令来简化许多复杂难打的命令
利用alias,可以将你要长期执行的命令,用一个你最喜欢的名字记下来, 用你最喜欢的编辑器打开.bashrc文件( 如$ vim ~/.bashrc) 在最后面输入: alias myssh='ss ...
- unity3d将C#打包成dll方法
方法一:用vs新建工程-C#库,添加UnityEngine.dll引用,注意.netframwork选3.5,编译C#脚本得到dll: 方法二:使用mono的mcs,具体如下 c#提供了dll打包,但 ...
- ffmpeg 编码(视屏)
分析ffmpeg_3.3.2 muxing 1:分析主函数,代码如下: int main(int argc, char **argv) { OutputStream video_st = { }, a ...
- 华为手机如何查看WiFi密码
2017.8.26 (补充) 博主更新华为手机系统之后,发现下面的教程已经不适用了,新系统在备份WLAN时强制要求设置密码,无法跳过,所以下面的教程仅适用于备份时可以跳过设置密码的系统. 有时候手机用 ...
- Python Flask 多环境配置
Python里取配置文件的时候,之前是使用的ini文件和python里configparser 模块: 可参考:https://www.cnblogs.com/feeland/p/4514771.ht ...