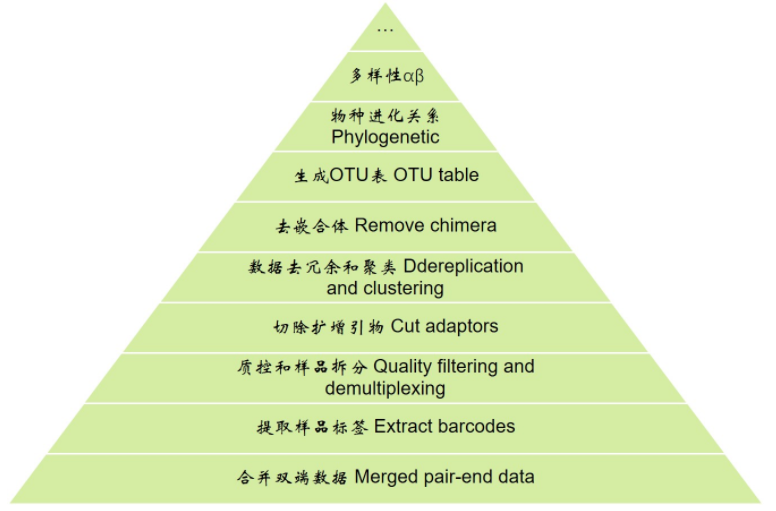
扩增子分析解读4去嵌合体 非细菌序列 生成代表性序列和OTU表
# 进入工作目录
cd example_PE250
# 下载Usearch推荐的参考数据库RDP
wget http://drive5.com/uchime/rdp_gold.fa
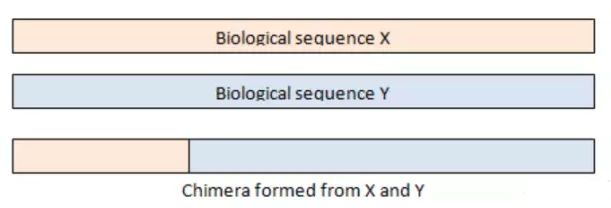
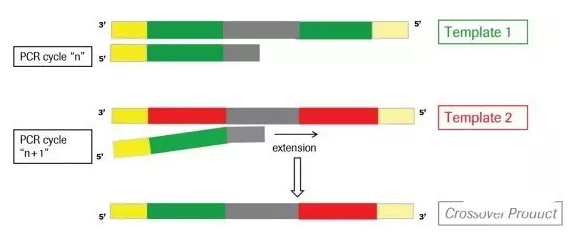
# 基于RDP数据库比对去除已知序列的嵌合体
./usearch10 -uchime2_ref temp/otus.fa \
-db rdp_gold.fa \
-chimeras temp/otus_chimeras.fa \
-notmatched temp/otus_rdp.fa \
-uchimeout temp/otus_rdp.uchime \
-strand plus -mode sensitive -threads 96
# 获得嵌合体的序列ID
grep '>' temp/otus_chimeras.fa | sed 's/>//g' > temp/otus_chimeras.id
# 剔除嵌合体的序列
filter_fasta.py -f temp/otus.fa -o temp/otus_non_chimera.fa -s temp/otus_chimeras.id -n
# 检查是否为预期的序列数量2820
grep '>' -c temp/otus_non_chimera.fa
# 下载Greengene最新数据库,320MB
wget -c ftp://greengenes.microbio.me/greengenes_release/gg_13_5/gg_13_8_otus.tar.gz
# 解压数据包后大小3.4G
tar xvzf gg_13_8_otus.tar.gz
# 将OTU与97%相似聚类的代表性序列多序列比对,大约8min
time align_seqs.py -i temp/otus_non_chimera.fa -t gg_13_8_otus/rep_set_aligned/97_otus.fasta -o temp/aligned/
# 无法比对细菌的数量
grep -c '>' temp/aligned/otus_non_chimera_failures.fasta # 1860
# 获得不像细菌的OTU ID
grep '>' temp/aligned/otus_non_chimera_failures.fasta|cut -f 1 -d ' '|sed 's/>//g' > temp/aligned/otus_non_chimera_failures.id
# 过滤非细菌序列
filter_fasta.py -f temp/otus_non_chimera.fa -o temp/otus_rdp_align.fa -s temp/aligned/otus_non_chimera_failures.id -n
# 看我们现在还有多少OTU:975
grep '>' -c temp/otus_rdp_align.fa
# 重命名OTU,这就是最终版的代表性序列,即Reference(可选,个人习惯)
awk 'BEGIN {n=1}; />/ {print ">OTU_" n; n++} !/>/ {print}' temp/otus_rdp_align.fa > result/rep_seqs.fa
# 生成OTU表
./usearch10 -usearch_global temp/seqs_usearch.fa -db result/rep_seqs.fa -otutabout temp/otu_table.txt -biomout temp/otu_table.biom -strand plus -id 0.97 -threads 10
# 结果信息 01:20 141Mb 100.0% Searching seqs_usearch.fa, 32.3% matched
# 默认10线程,用时1分20秒,有32.3%的序列匹配到OTU上;用30线程反而用时3分04秒,不是线程越多越快,分发任务也是很费时间的
扩增子分析解读4去嵌合体 非细菌序列 生成代表性序列和OTU表的更多相关文章
- 扩增子分析解读5物种注释 OTU表操作
本节课程,需要先完成<扩增子分析解读>系列之前的操作 1质控 实验设计 双端序列合并 2提取barcode 质控及样品拆分 切除扩增引物 3格式转换 去冗余 聚类 4去嵌合体 非细菌序列 ...
- 扩增子分析解读2提取barcode 质控及样品拆分 切除扩增引物
本节课程,需要完成扩增子分析解读1质控 实验设计 双端序列合并 先看一下扩增子分析的整体流程,从下向上逐层分析 分析前准备 # 进入工作目录 cd example_PE250 上一节回顾:我们拿到了双 ...
- 扩增子分析解读6进化树 Alpha Beta多样性
分析前准备 # 进入工作目录 cd example_PE250 上一节回顾:我们的OTU获得了物种注释,并学习OTU表的各种操作————添加信息,格式转换,筛选信息. 接下来我们学习对OTU序列的 ...
- 扩增子图表解读6韦恩图:比较组间共有和特有OTU或分类单元
韦恩图 Venn Diagram Venn Diagram,也称韦恩图.维恩图.文氏图,用于显示元素集合重叠区域的图示. 韦图绘制工具 常用R语言的VennDiagram包绘制,输出PDF格式方便 ...
- 解读人:谭亦凡,Macrophage phosphoproteome analysis reveals MINCLE-dependent and -independent mycobacterial cord factor signaling(巨噬细胞磷酸化蛋白组学分析揭示MINCLE依赖和非依赖的分支杆菌索状因子信号通路)(MCP换)
发表时间:2019年4月 IF:5.232 一. 概述: 分支杆菌索状因子TDM(trehalose-6,6’-dimycolate)能够与巨噬细胞C-型凝集素受体(CLR)MINCLE结合引起下游通 ...
- 扩增子图表解读5火山图:差异OTU的数量及变化规律
火山图 Volcano plot 在统计学上,火山图是一种类型的散点图,被用于在大数据中快速鉴定变化.由于它的形成像火山喷发的样子,所以被称为火山图.和上文讲的曼哈顿图类似. 火山图基本元素 火山 ...
- 扩增子图表解读1箱线图:Alpha多样性
箱线图 箱形图(Box-plot)又称为盒须图.盒式图或箱线图,是一种用作显示一组数据分散情况资料的统计图.因形状如箱子而得名.在宏基因组领域,常用于展示样品组中各样品Alpha多样性的分布 第一种情 ...
- 如何分析解读systemstat dump产生的trc文件
ORACLE数据库的systemstat dump生成trace文件虽然比较简单,但是怎么从trace文件中浩如烟海的信息中提炼有用信息,并作出分析诊断是一件技术活,下面收集.整理如何分析解读syst ...
- 扩增子分析QIIME2. 1简介和安装
原网站:https://blog.csdn.net/woodcorpse/article/details/75103929 声明:本文为QIIME2官方帮助文档的中文版,由中科院遗传发育所刘永鑫博士翻 ...
随机推荐
- 两种常见的UITabBarController+UINavigationController模式分析比较
绝大部分软件都采用了UITabBarController+UINavigationController的设计模式,这是一种很主流很经典的设计方式,而另外一种UINavigationController ...
- iOS单例设计模式具体解说(单例设计模式不断完好的过程)
在iOS中有非常多的设计模式,有一本书<Elements of Reusable Object-Oriented Software>(中文名字为<设计模式>)讲述了23种软件设 ...
- Linux 编译C++ 与 设置 Vim
1. Linux 下编译c++ vim test.cpp // 创建文件 g++ test.cpp // 编译文件 ./a.out // 执行文件 g++ test.cpp ...
- Codeforces(429D - Tricky Function)近期点对问题
D. Tricky Function time limit per test 2 seconds memory limit per test 256 megabytes input standard ...
- cssTest
html <!doctype html> <html> <head> <meta charset="utf-8"> <meta ...
- eclipse android开发,文本编辑xml文件,给控件添加ID后,R.java,不自动的问题。
直接编辑xml文件给控件添加id,不自动更新.原来的id写法:@id/et_tel 然后改写成这样:@+id/et_tel 然后就好了!操`1
- data-toggle data-target
data-toggle https://stackoverflow.com/questions/30629974/how-does-the-data-toggle-attribute-work-wha ...
- BSGS算法及拓展
https://www.zybuluo.com/ysner/note/1299836 定义 一种用来求解高次同余方程的算法. 一般问题形式:求使得\(y^x\equiv z(mod\ p)\)的最小非 ...
- 基于.Net Core的API框架的搭建(4)
6.加入日志功能 日志我们选用log4net,首先引入程序包: 选择2.0.8版本安装.然后在项目根目录新增log4net的配置文件log4net.config: <?xml version=& ...
- 字符类型C++(ascll码表)
ascll码: 序号 字符 序号 字符 序号 字符 序号 字符 序号 字符 序号 字符 32 空格 48 0 64 @ 80 P 96 ` 112 p 33 ! 49 1 65 A 81 Q 97 a ...