单细胞分析实录(9): 展示marker基因的4种图形(二)
在上一篇中,我已经讲解了展示marker基因的前两种图形,分别是tsne/umap图、热图,感兴趣的读者可以回顾一下。这一节我们继续学习堆叠小提琴图和气泡图。
3. 堆叠小提琴图展示marker基因
相比于其他可视化形式,小提琴图可以更直观地展示某一类亚群的某一个基因的表达分布情况。我的marker基因一共选了12个,下面来画图:
Seurat内置的VlnPlot函数可以直接画,
library(xlsx)
markerdf2=read.xlsx("ref_marker2.xlsx",sheetIndex = 1)
markerdf2$gene=as.character(markerdf2$gene)
mye.seu=readRDS("mye.seu.rds")
mye.seu$celltype=factor(mye.seu$celltype,levels = sort(unique(mye.seu$celltype)))
Idents(mye.seu)="celltype"
VlnPlot(mye.seu, features = markerdf2$gene, pt.size = 0, ncol = 1)+
scale_x_discrete("")+
theme(
axis.text.x.bottom = element_blank()
)
ggsave("vln1.pdf",width = 20,height = 80,units = "cm")
其中pt.size参数表示点的大小,一个点就是一个细胞,一般可以直接设置为0,即不显示点,只画小提琴,看上去更加清楚。尽管此处我对标度和主题进行了调整,但我发现这只对单个feature有用,多个feature时就不起作用了,后续就用AI来简单编辑一下吧。
需要注意的是,图的颜色是根据亚群的类别来划分的,并不是根据基因来区分。
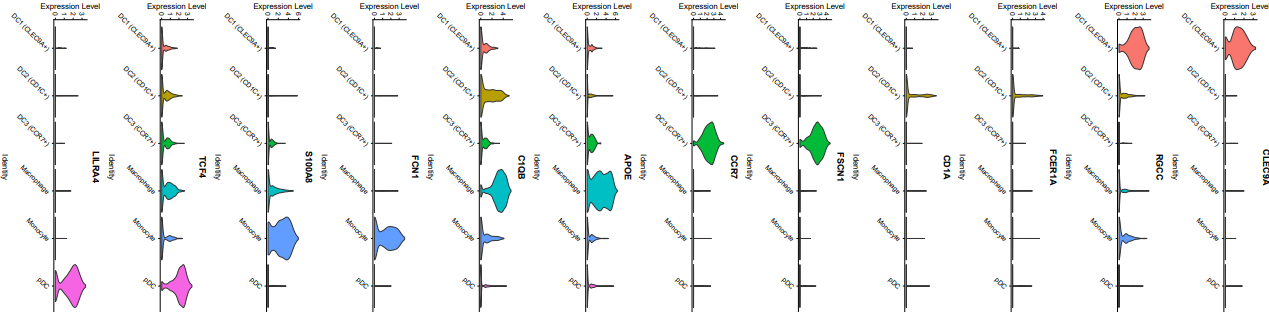
第二种方法,ggplot2代码如下
library(reshape2)
vln.df=as.data.frame(mye.seu[["RNA"]]@data[markerdf2$gene,])
vln.df$gene=rownames(vln.df)
vln.df=melt(vln.df,id="gene")
colnames(vln.df)[c(2,3)]=c("CB","exp")
#数据格式如下
# > head(vln.df)
# gene CB exp
# 1 CLEC9A N01_AAACGGGCATTTCAGG_1 0.000
# 2 RGCC N01_AAACGGGCATTTCAGG_1 0.000
# 3 FCER1A N01_AAACGGGCATTTCAGG_1 0.000
# 4 CD1A N01_AAACGGGCATTTCAGG_1 0.000
# 5 FSCN1 N01_AAACGGGCATTTCAGG_1 1.104
# 6 CCR7 N01_AAACGGGCATTTCAGG_1 0.000
anno=mye.seu@meta.data[,c("CB","celltype")]
vln.df=inner_join(vln.df,anno,by="CB")
vln.df$gene=factor(vln.df$gene,levels = markerdf2$gene) #为了控制画图的基因顺序
vln.df%>%ggplot(aes(celltype,exp))+geom_violin(aes(fill=gene),scale = "width")+
facet_grid(vln.df$gene~.,scales = "free_y")+
scale_fill_brewer(palette = "Set3",direction = 1)+
scale_x_discrete("")+scale_y_continuous("")+
theme_bw()+
theme(
axis.text.x.bottom = element_text(angle = 45,hjust = 1,vjust = 1),
panel.grid.major = element_blank(),panel.grid.minor = element_blank(),
legend.position = "none"
)
ggsave("vln2.pdf",width = 11,height = 22,units = "cm")
geom_violin()函数的scale参数为"width"时,所有小提琴有相同的宽度,默认是"area",有相同的面积;facet_grid()用来分面,文中用的是多行一列,scales = "free_y"表示不同行之间可以有不同范围的y值;scale_fill_brewer()使用ColorBrewer调色板。
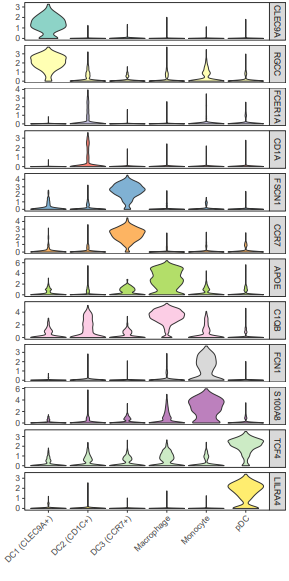
这个图的颜色根据基因来区分,有时可能还会看到小提琴图的颜色是用亚群某个基因的表达均值来映射的,比如
vln.df$celltype_gene=paste(vln.df$celltype,vln.df$gene,sep = "_")
stat.df=as.data.frame(vln.df%>%group_by(celltype,gene)%>%summarize(mean=mean(exp)))
stat.df$celltype_gene=paste(stat.df$celltype,stat.df$gene,sep = "_")
stat.df=stat.df[,c("mean","celltype_gene")]
vln.df=inner_join(vln.df,stat.df,by="celltype_gene")
vln.df$mean=ifelse(vln.df$mean > 3, 3, vln.df$mean)
#这里的阈值3要提前综合所有基因看一下
vln.df%>%ggplot(aes(celltype,exp))+geom_violin(aes(fill=mean),scale = "width")+
facet_grid(vln.df$gene~.,scales = "free_y")+
scale_fill_gradient(limits=c(0,3),low = "lightgrey",high = "yellow")+
scale_x_discrete("")+scale_y_continuous("",expand = c(0.02,0))+
theme_bw()+
theme(
panel.grid.major = element_blank(),panel.grid.minor = element_blank(),
axis.text.x.bottom = element_text(angle = 45,hjust = 1,vjust = 1)
)
ggsave("vln3.pdf",width = 11,height = 22,units = "cm")
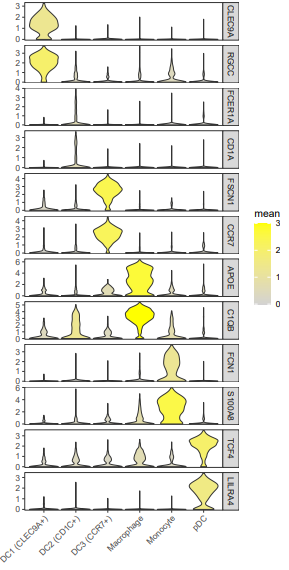
4. 气泡图展示marker基因
Seurat的画法是这样的,
DotPlot(mye.seu, features = markerdf2$gene)+RotatedAxis()+
scale_x_discrete("")+scale_y_discrete("")
#其余的微调同ggplot2
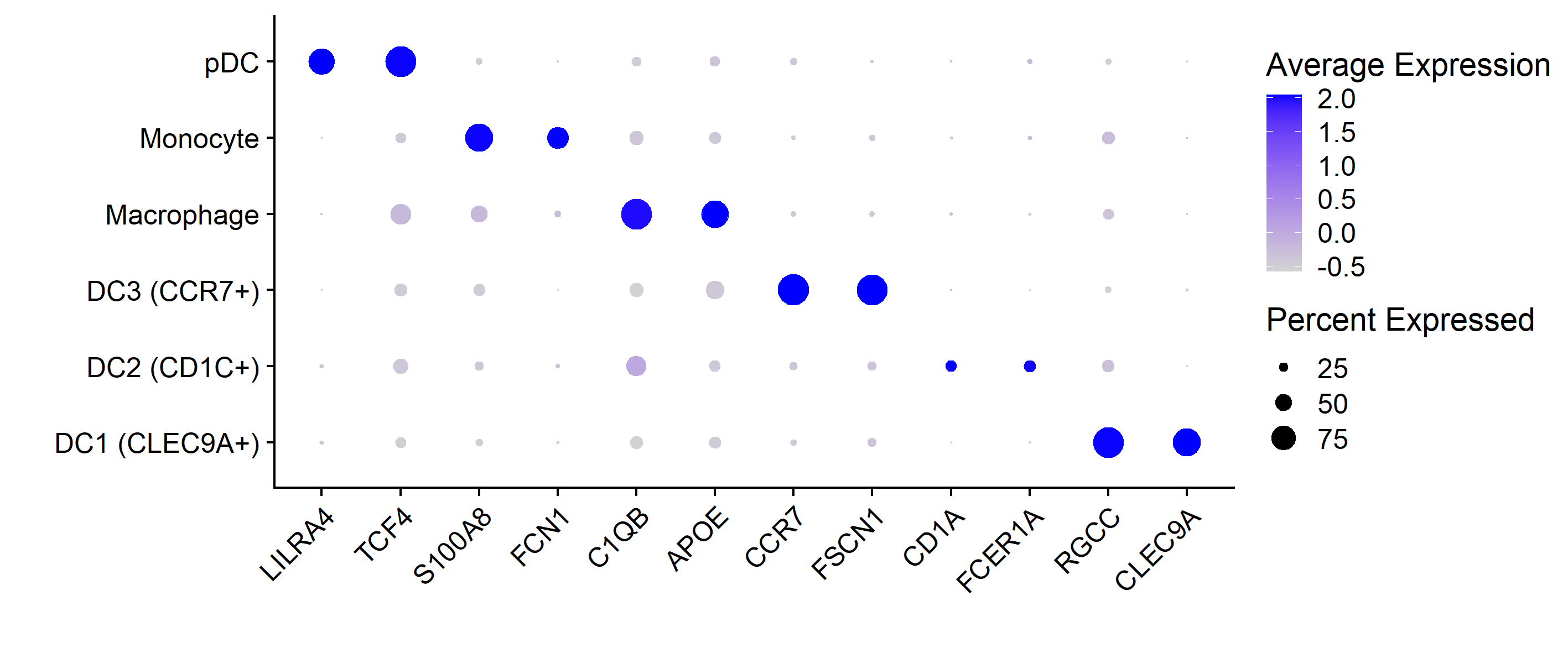
第二种方法,ggplot2代码如下
bubble.df=as.matrix(mye.seu[["RNA"]]@data[markerdf2$gene,])
bubble.df=t(bubble.df)
bubble.df=as.data.frame(scale(bubble.df))
bubble.df$CB=rownames(bubble.df)
bubble.df=merge(bubble.df,mye.seu@meta.data[,c("CB","celltype")],by = "CB")
bubble.df$CB=NULL
celltype_v=c()
gene_v=c()
mean_v=c()
ratio_v=c()
for (i in unique(bubble.df$celltype)) {
bubble.df_small=bubble.df%>%filter(celltype==i)
for (j in markerdf2$gene) {
exp_mean=mean(bubble.df_small[,j])
exp_ratio=sum(bubble.df_small[,j] > min(bubble.df_small[,j])) / length(bubble.df_small[,j])
celltype_v=append(celltype_v,i)
gene_v=append(gene_v,j)
mean_v=append(mean_v,exp_mean)
ratio_v=append(ratio_v,exp_ratio)
}
}
plotdf=data.frame(
celltype=celltype_v,
gene=gene_v,
exp=mean_v,
ratio=ratio_v
)
plotdf$celltype=factor(plotdf$celltype,levels = sort(unique(plotdf$celltype)))
plotdf$gene=factor(plotdf$gene,levels = rev(as.character(markerdf2$gene)))
plotdf$exp=ifelse(plotdf$exp>3,3,plotdf$exp)
plotdf%>%ggplot(aes(x=celltype,y=gene,size=ratio,color=exp))+geom_point()+
scale_x_discrete("")+scale_y_discrete("")+
scale_color_gradientn(colours = rev(c("#FFD92F","#FEE391",brewer.pal(11, "Spectral")[7:11])))+
scale_size_continuous(limits = c(0,1))+theme_bw()+
theme(
axis.text.x.bottom = element_text(hjust = 1, vjust = 1, angle = 45)
)
ggsave(filename = "bubble2.pdf",width = 9,height = 12,units = c("cm"))
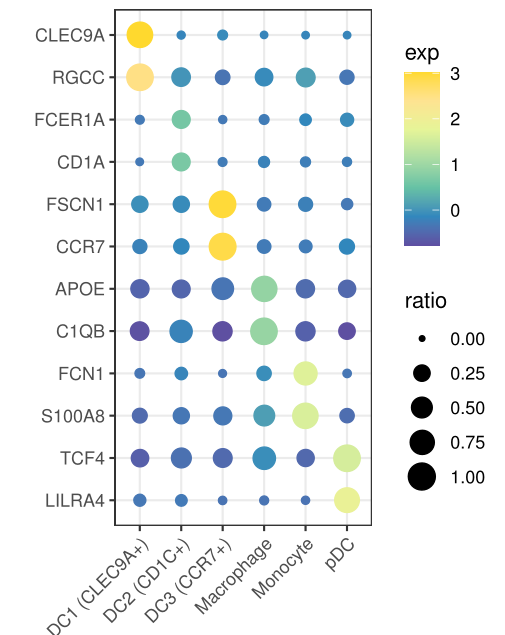
这两种方法具体函数定义略有差异,所以气泡图看上去不太一样
到这里,marker基因的可视化就结束了,基本就是这些。如果你觉得上述内容对你有用,欢迎转发,点赞!有任何疑问可以在公众号后台提出,我都会回复的。
因水平有限,有错误的地方,欢迎批评指正!
单细胞分析实录(9): 展示marker基因的4种图形(二)的更多相关文章
- 单细胞分析实录(8): 展示marker基因的4种图形(一)
今天的内容讲讲单细胞文章中经常出现的展示细胞marker的图:tsne/umap图.热图.堆叠小提琴图.气泡图,每个图我都会用两种方法绘制. 使用的数据来自文献:Single-cell transcr ...
- 【代码更新】单细胞分析实录(20): 将多个样本的CNV定位到染色体臂,并画热图
之前写过三篇和CNV相关的帖子,如果你做肿瘤单细胞转录组,大概率看过: 单细胞分析实录(11): inferCNV的基本用法 单细胞分析实录(12): 如何推断肿瘤细胞 单细胞分析实录(13): in ...
- 【代码更新】单细胞分析实录(21): 非负矩阵分解(NMF)的R代码实现,只需两步,啥图都有
1. 起因 之前的代码(单细胞分析实录(17): 非负矩阵分解(NMF)代码演示)没有涉及到python语法,只有4个python命令行,就跟Linux下面的ls grep一样的.然鹅,有几个小伙伴不 ...
- 单细胞分析实录(5): Seurat标准流程
前面我们已经学习了单细胞转录组分析的:使用Cell Ranger得到表达矩阵和doublet检测,今天我们开始Seurat标准流程的学习.这一部分的内容,网上有很多帖子,基本上都是把Seurat官网P ...
- 单细胞分析实录(4): doublet检测
最近Cell Systems杂志发表了一篇针对现有几种检测单细胞测序doublet的工具的评估文章,系统比较了常见的例如Scrublet.DoubletFinder等工具在检测准确性.计算效率等方面的 ...
- 单细胞分析实录(3): Cell Hashing数据拆分
在之前的文章里,我主要讲了如下两个内容:(1) 认识Cell Hashing:(2): 使用Cell Ranger得到表达矩阵.相信大家已经知道了cell hashing与普通10X转录组的差异,以及 ...
- 单细胞分析实录(18): 基于CellPhoneDB的细胞通讯分析及可视化 (上篇)
细胞通讯分析可以给我们一些细胞类群之间相互调控/交流的信息,这种细胞之间的调控主要是通过受配体结合,传递信号来实现的.不同的分化.疾病过程,可能存在特异的细胞通讯关系,因此阐明这些通讯关系至关重要. ...
- 单细胞分析实录(19): 基于CellPhoneDB的细胞通讯分析及可视化 (下篇)
在上一篇帖子中,我介绍了CellPhoneDB的原理.实际操作,以及一些值得注意的地方.这一篇继续细胞通讯分析的可视化. 公众号后台回复20210723获取本次演示的测试数据,以及主要的可视化代码. ...
- 单细胞分析实录(2): 使用Cell Ranger得到表达矩阵
Cell Ranger是一个"傻瓜"软件,你只需提供原始的fastq文件,它就会返回feature-barcode表达矩阵.为啥不说是gene-cell,举个例子,cell has ...
随机推荐
- Codeforces Round #671 (Div. 2)
比赛链接:https://codeforces.com/contest/1419 A. Digit Game 题意 给出一个 $n$ 位数,游戏规则如下: 1-indexed Raze标记奇数位 Br ...
- Codeforces Round #647 (Div. 2) B. Johnny and His Hobbies(枚举)
题目链接:https://codeforces.com/contest/1362/problem/B 题意 有一个大小及元素值均不超过 $1024$ 的正整数集合,求最小正整数 $k$,使得集合中的每 ...
- Codeforces Round #665 (Div. 2) Distance and Axis、
题目链接:Distance and Axis 题意:在ox轴上,给出点A的横坐标x,你可以向左或右移动点A(x+1/x-1),问你最小移动A的次数,以使得可以在ox轴上找到B点位置,B点满足从O到B的 ...
- G - Can you answer these queries? & N - 花神游历各国
A lot of battleships of evil are arranged in a line before the battle. Our commander decides to us ...
- AtCoder Beginner Contest 188 C - ABC Tournament (模拟)
题意:有\(2^n\)个人站成一排比赛,刚开始每个人都和自己右边的人进行比赛,赢得人晋级下一轮(下标的小的在前面),不断重复这个过程,问最后拿到第二名的人的编号. 题解:根据题意,可以用vector直 ...
- 对模拟器虚假设备识别能力提升15%!每日清理大师App集成系统完整性检测
前言 每日清理大师是一款智能便捷的手机清理软件,可快速清理无用缓存.垃圾文件和应用残留,还可深度清理如社交软件中的无用缓存等,有效解决手机卡顿.耗电快.内存不足等问题.每日清理大师App在结合了系统完 ...
- Redis Cluster 分布式集群(上)
Redis Cluster 介绍 Redis 集群是一个可以在多个Redis节点之间进行数据共享的设施(installation): Redis 集群不支持那些需要同时处理多个键的 Redis 命令, ...
- hdu5303贪心
http://acm.hdu.edu.cn/showproblem.php?pid=5303 说一下题目大意.. 有一个长为L的环..你家在原点位置0,那么剩下L-1个点上种有一些树, 给你树的位置和 ...
- 5种设置ASP.NET Core应用程序URL的方法
默认情况下,ASP.NET Core应用程序监听以下URL: http://localhost:5000 https://localhost:5001 在这篇文章中,我展示了5种不同的方式来更改您的应 ...
- Object Destructuring Assignment vs Object.assign
Object Destructuring Assignment vs Object.assign // const params = Object.assign({}, this.$route.par ...