测序数据质控-FastQC
通常我们下机得到的数据是raw reads,但是公司通常会质控一份给我们,所以到很多人手上就是clean data了。我们再次使用fastqc来进行测序数据质量查看以及结果分析。
fastqc的操作:
--noextract:结果文件压缩 [默认]
-c --contaminants:指定污染序列。文件格式 name[tab]sequence
-a --adapters:指定接头序列。文件格式name[tab]sequence
-k --kmers:指定kmers长度(2-10bp,默认7bp)
-q --quiet: 安静模式-不显示进展
# 不显示此类进展
Approx 55% complete for ERR522959_2.fastq
Approx 60% complete for ERR522959_2.fastq
Approx 65% complete for ERR522959_2.fastq
Approx 70% complete for ERR522959_2.fastq
1.下载并安装fastqc
wget http://www.bioinformatics.babraham.ac.uk/projects/fastqc/fastqc_v0.11.8.zip
unzip fastqc_v0.11.8.zip
cd FastQC
chmod 755 fastqc
2.下载测序数据
数据来源:单细胞RNA-seq,SMART-seq2. “a single cell from an mESC dataset produced by (Kolodziejczyk et al. 2015). The cells were sequenced using the SMART-seq2 library preparation protocol and the reads are paired end.”
wget ftp://ftp.sra.ebi.ac.uk/vol1/fastq/ERR522/ERR522959/ERR/ERR522959_1.fastq.gz
wget ftp://ftp.sra.ebi.ac.uk/vol1/fastq/ERR522/ERR522959/ERR/ERR522959_1.fastq.gz
3. 运行fastqc
nohup ~/software/FastQC/fastqc -f fastq -o result/ ERR522959_1.fastq ERR522959_2.fastq &
4. 结果文件分析

zip文件包含了html文件,里面有数据质量的图片。
我们来查看html文件

各种颜色是各项标准分析结果:绿色代表"PASS";
黄色代表"WARN";红色代表"FAIL"。
4.1 Basic Statics
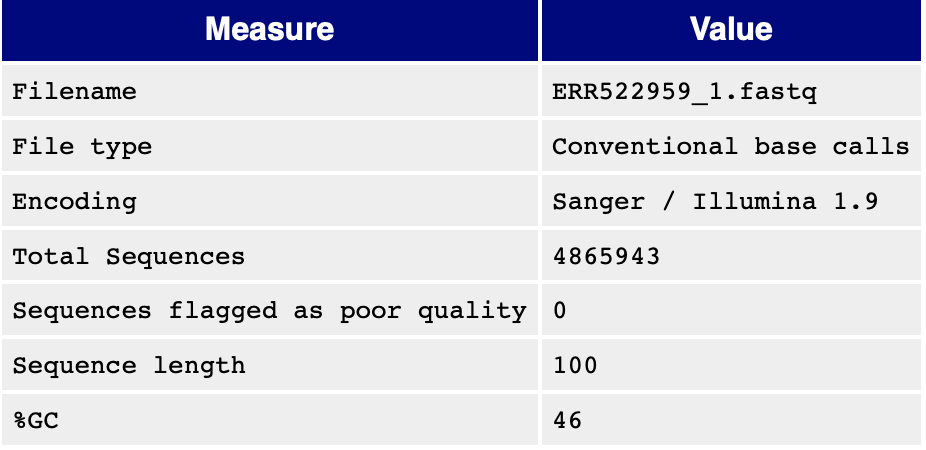
一些基本信息在这个表格,其中的Encoding 很重要,表示测序数据的编码方式。编码方式有很多种:

这里的Illumina1.9 就是 Phred+33。
4.2 每条reads不同位置(1-100bp)的测序质量统计
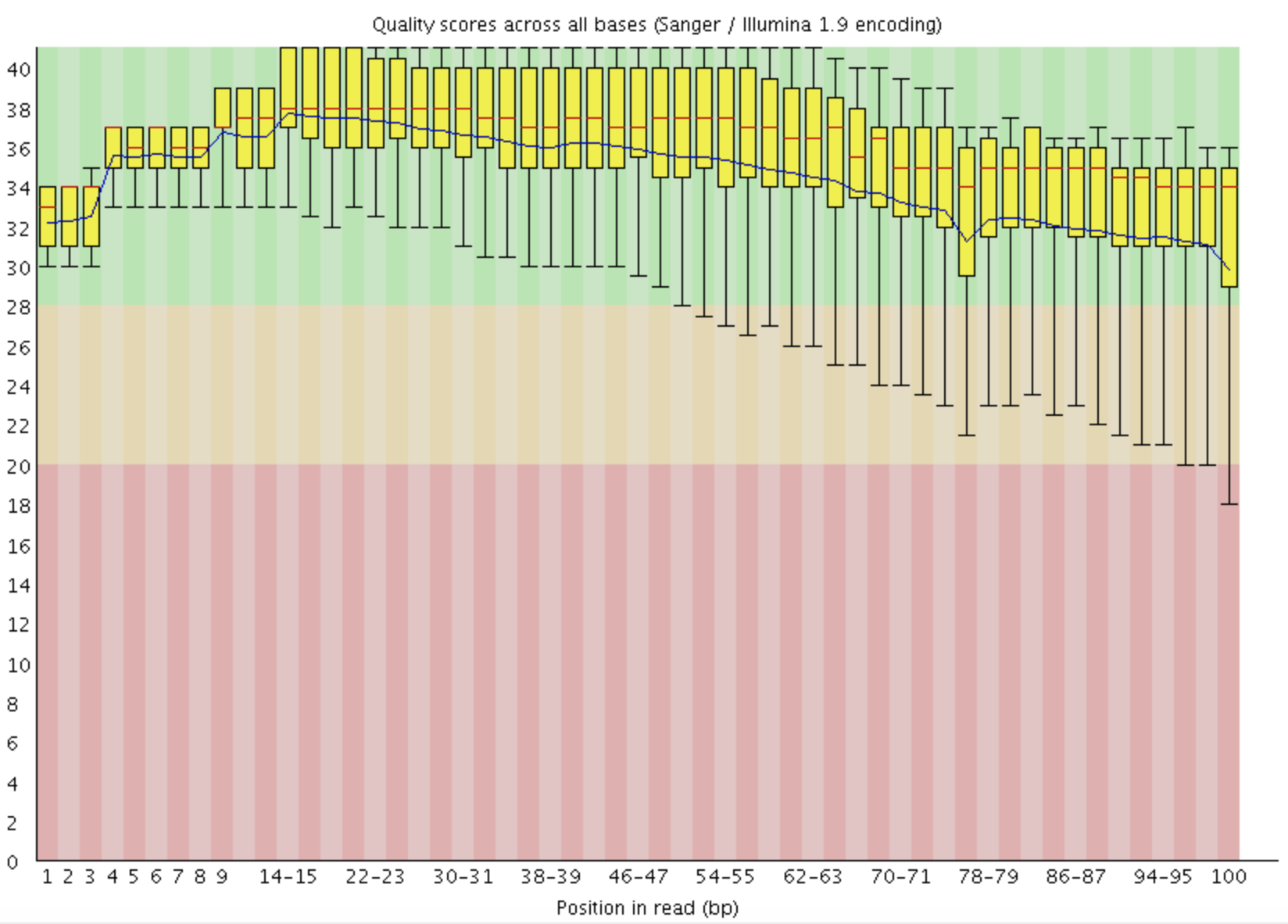
这是一个boxplot,注意看横坐标1-100bp的表示方式。一首一尾是单bp的,中间部分是2bp一个单位。Y轴是测序的质量Q=Phred=-log10(error rate),所以Q=20时,错误率为0.01,就是99%的正确率(两个9,Q20)。Q=30, 错误率就是0.001, 就是99.9%的正确率(三个9,Q30),通常测序合同对Q20,Q30有指标要求。红色表示中位数,黄色是25%-75%区间,触须是10%-90%区间,蓝线是平均数。平均每个碱基的测序质量boxplot下四分位线在30分以上,则认为测序质量非常好;一般情况下,reads首尾质量较差。若任一位置的下四分位数低于10或中位数低于25,报"WARN";若任一位置的下四分位数低于5或中位数低于20,报"FAIL"。
4.3
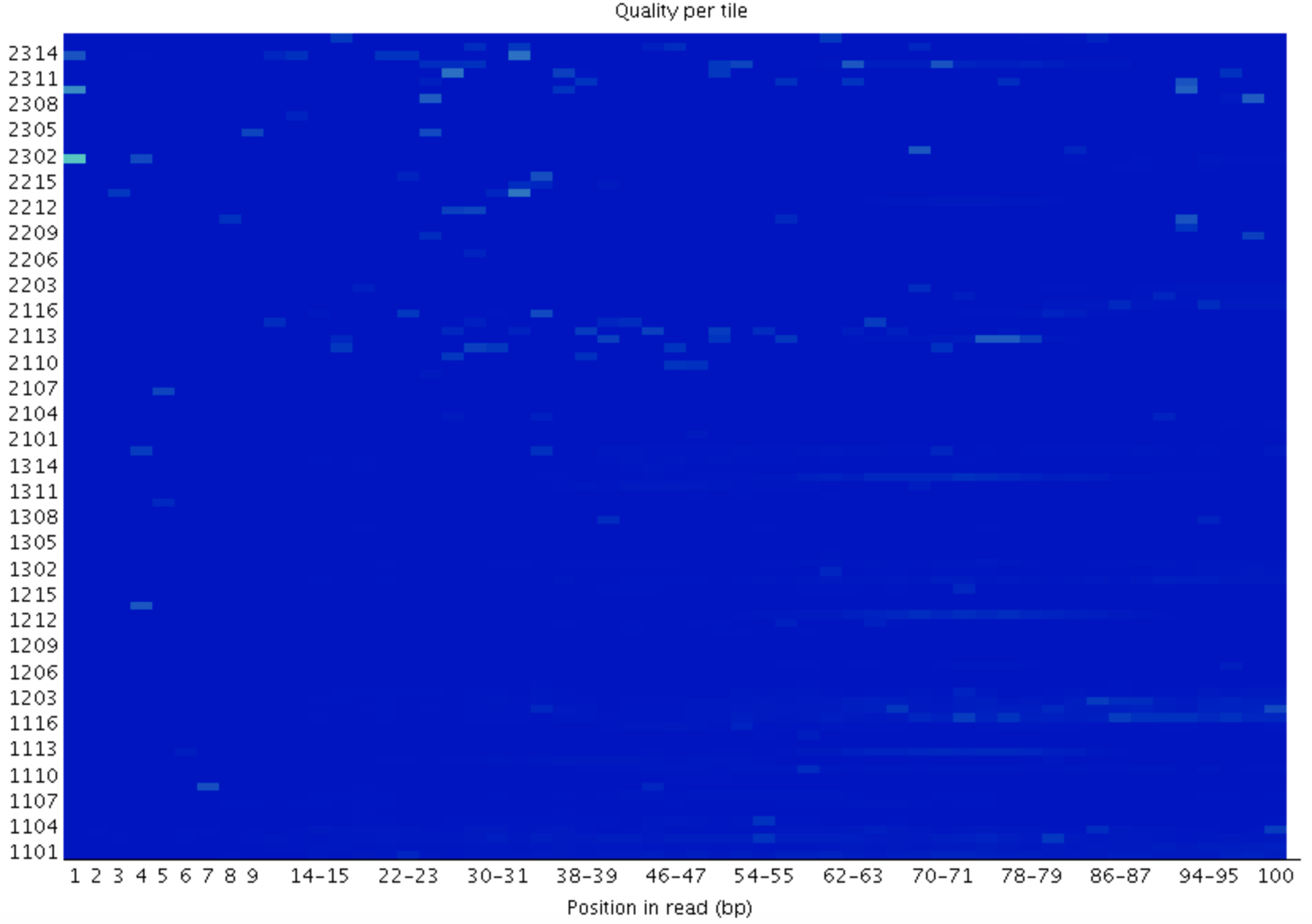
检查reads中每一个碱基位置在不同的测序小孔之间的偏离度,蓝色表示低于平均偏离度,偏离度小,质量好;越红表示偏离平均质量越多,质量也越差。如果出现质量问题可能是短暂的,如有气泡产生,也可能是长期的,如在某一小孔中存在残骸,问题不大。

横轴为序列测序质量,纵轴是reads数目。一般认为90%的reads测序质量在35分以上,则认为该测序质量非常好。
当测序质量峰值小于27(错误率0.2%)时报"WARN";
当峰值小于20(错误率1%)时报"FAIL"。
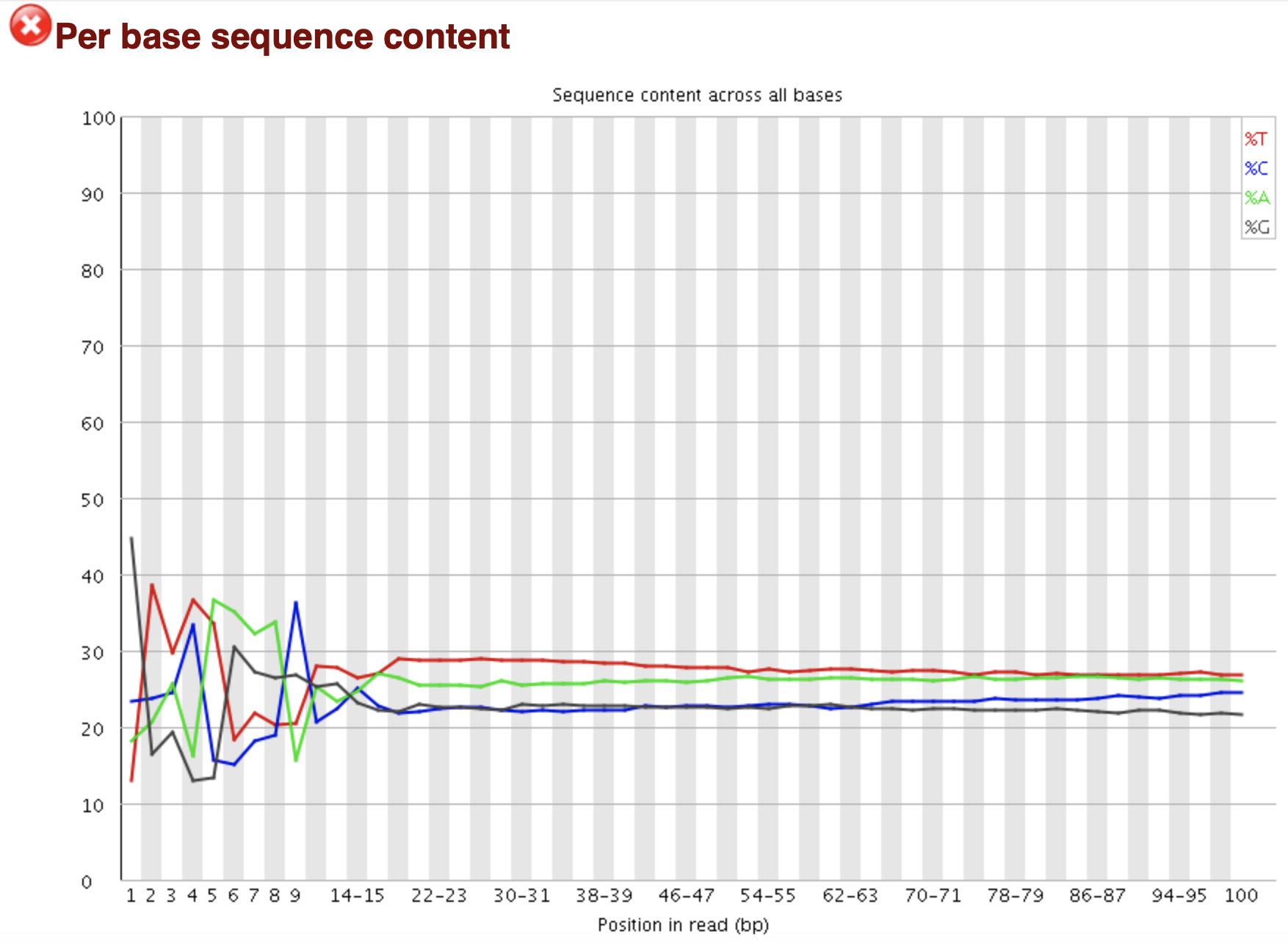
横轴为碱基位置,纵轴为百分比。因为随机的文库中,正常情况下所有位置出现某种碱基的概率是相近的,因此好的测序结果中四条线应该平行且接近。当部分位置碱基的比例出现bias时,即四条线在某些位置纷乱交织,往往提示我们有overrepresented sequence的污染。当所有位置的碱基比例一致的表现出bias时,即四条线平行但分开,往往代表文库有bias (建库过程或本身特点),或者是测序中的系统误差。
当任一位置的A/T比例与G/C比例相差超过10%,报"WARN";
当任一位置的A/T比例与G/C比例相差超过20%,报"FAIL"。
 统计reads的平均GC含量分布
统计reads的平均GC含量分布
红线是实际情况,蓝线是理论分布(正态分布,均值不一定在50%,而是由平均GC含量推断的)。 曲线形状的偏差往往是由于文库的污染或是部分reads构成的子集有偏差(overrepresented reads)。形状接近正态但偏离理论分布的情况提示我们可能有系统偏差。
偏离理论分布的reads超过15%时,报"WARN";偏离理论分布的reads超过30%时,报"FAIL"。

reads某个位置无法确定是何种碱基时,使用N代替;
正常情况下,N的比例是很小的,所以图上常常看到一条直线,但放大Y轴之后会发现还是有N的存在,这不算问题。当Y轴在0%-100%的范围内也能看到“鼓包”时,说明测序系统出了问题。
当任意位置的N的比例超过5%,报"WARN";
当任意位置的N的比例超过20%,报"FAIL"。
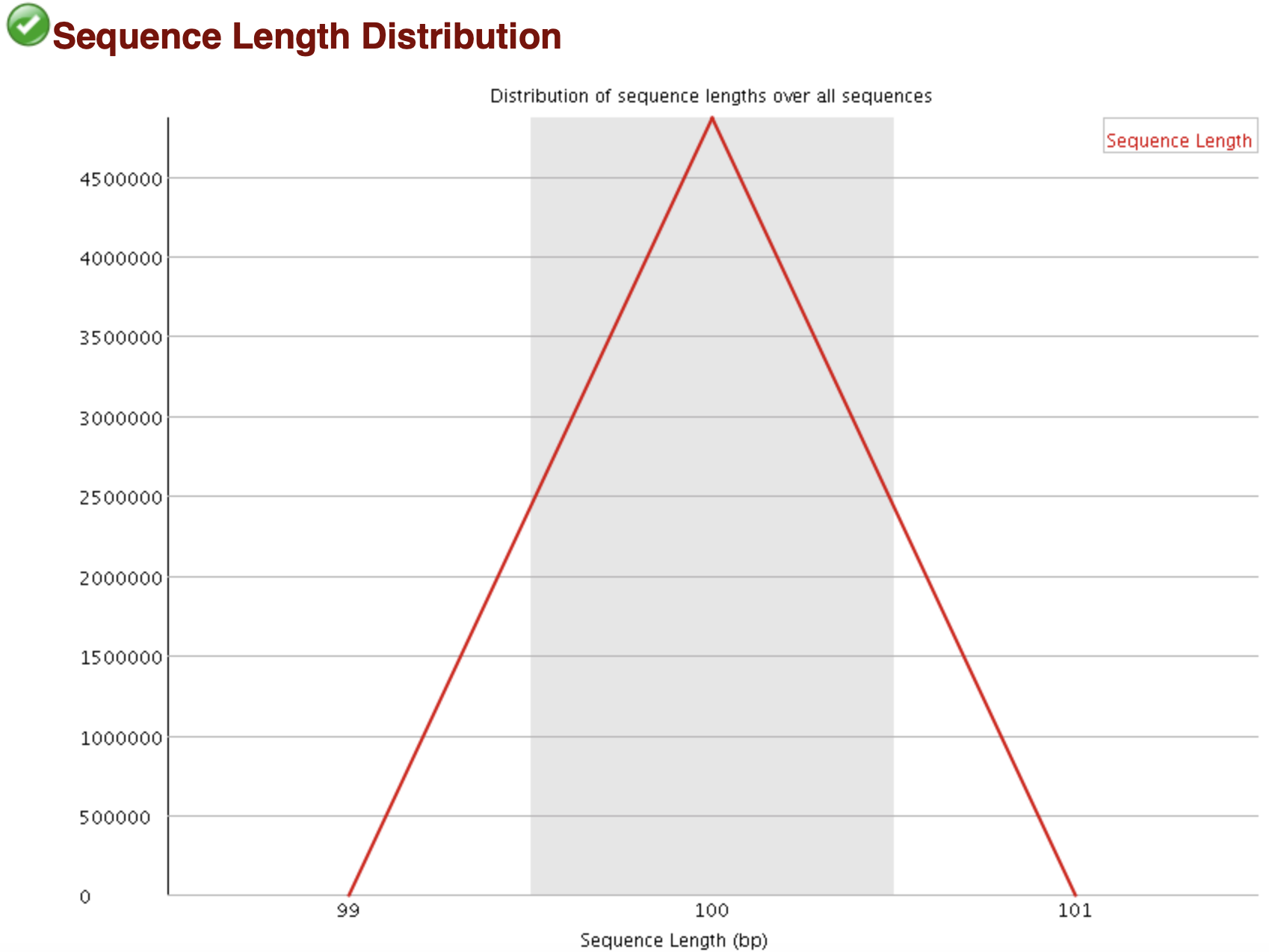
reads长度分布
为了防止建库或者测序时有一些不规则长度的序列也被进行测序而进行的一个对长度的统计,当所有序列的长度不一样,fastqc就会警告。
当reads长度不一致时报"WARN";
当有长度为0的read时报“FAIL”。
统计序列完全一样的reads的频率
统计reads重复水平测序本身就会产生重复reads,测序深度越高,reads重复数越大;如果重复出现峰值,就提示可能b存在偏差(如建库过程中的PCR duplication)。
横坐标是重复的次数,纵坐标是duplicated reads占unique reads种数百分比。
fastqc抽取reads文件前200,000条reads统计其重复情况。重复数目大于等于10的reads被合并统计,这也是为什么我们看到上图的最右侧略有上扬。大于75bp的reads只取50bp进行比较。由于reads越长错误率越高,所以其重复程度仍有可能被低估。
当非unique的reads占总数的比例大于20%时,报"WARN";
当非unique的reads占总数的比例大于50%时,报"FAIL“。
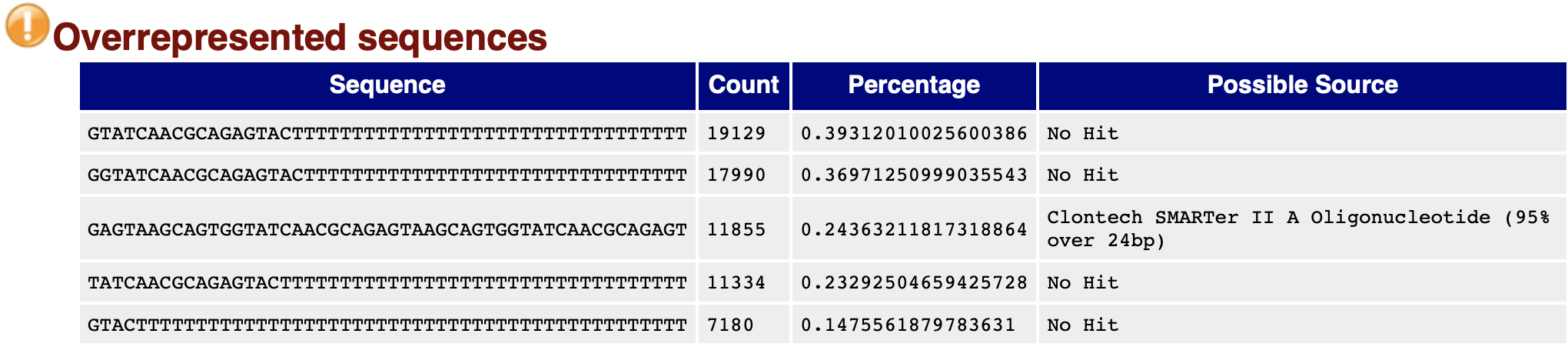
过度重复出现的序列的统计信息
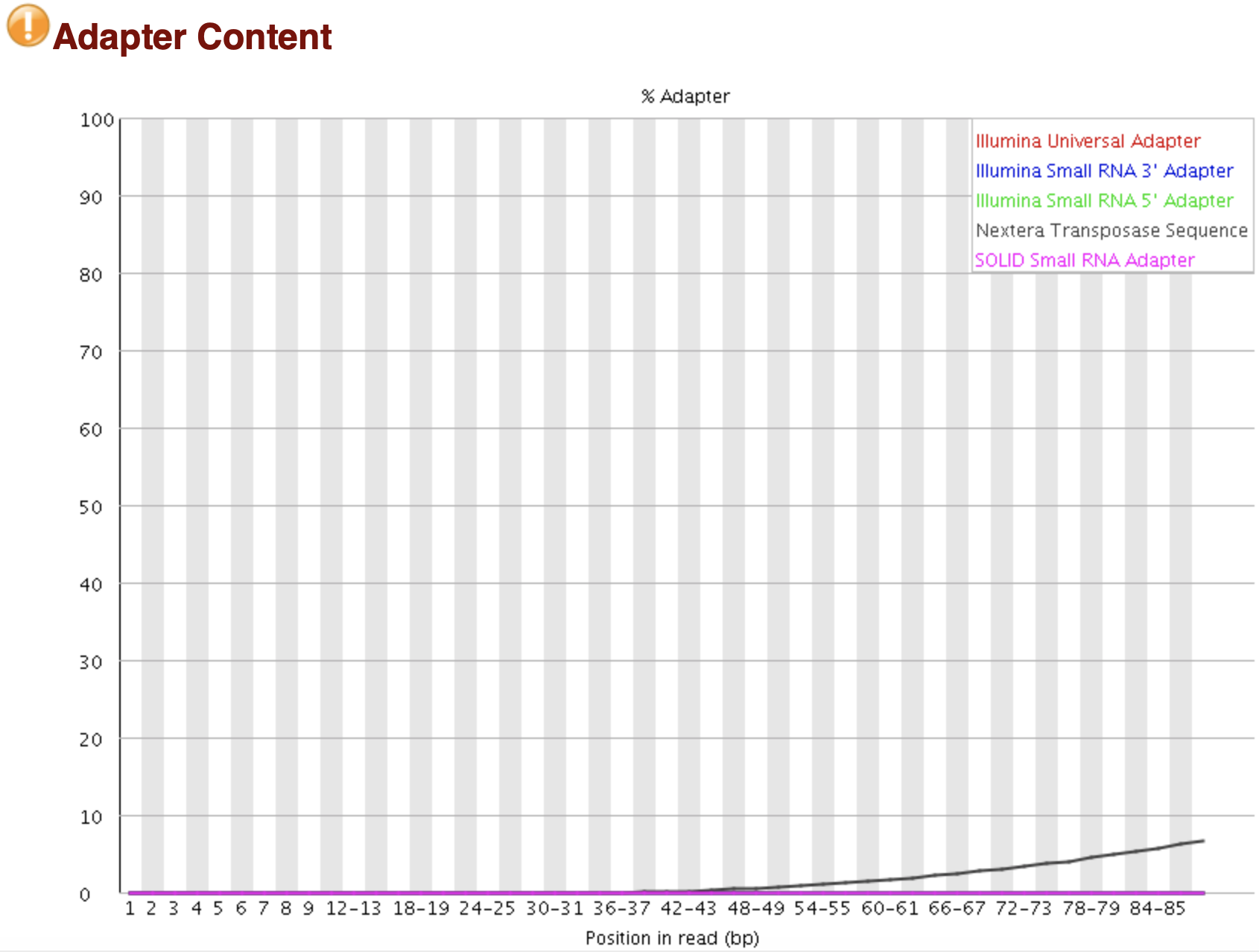
Adapter序列在reads中出现概率
接头序列统计,>5%时是Warning,>10%时是Failure。
一般测序read的长度大于插入片段(待测序列)的长度时会发生。对于WGS建库测序来讲,一般不会发生,因为他的待测序列要几百bp,而测序也就150bp算高了。但是对于RNA-seq,一般测序序列比较短,尤其是miRNA,只有几十bp,这是就会测到read末尾的接头。
本文图片由fastqc生成,后面对图描述的文字来源于简书:
“作者:_eason_
链接:https://www.jianshu.com/p/835fd925d6ee
來源:简书”

测序数据质控-FastQC的更多相关文章
- 【转录组入门】3:了解fastq测序数据
操作:需要用安装好的sratoolkit把sra文件转换为fastq格式的测序文件,并且用fastqc软件测试测序文件的质量 作业:理解测序reads,GC含量,质量值,接头,index,fastqc ...
- 弗雷塞斯 从生物学到生物信息学到机器学习 转录组入门(3):了解fastq测序数据
sra文件转换为fastq格式 1 fastq-dump -h --split-3 也就是说如果SRA文件中只有一个文件,那么这个参数就会被忽略.如果原文件中有两个文件,那么它就会把成对的文件按*_1 ...
- 转录组入门(3):了解fastq测序数据
sra文件转换为fastq格式 fastq-dump -h --split-3 也就是说如果SRA文件中只有一个文件,那么这个参数就会被忽略.如果原文件中有两个文件,那么它就会把成对的文件按*_1.f ...
- fastx_toolkit去除测序数据中的接头和低质量的reads
高通量测序数据下机后得到了fastq的raw_data,通常测序公司在将数据返还给客户之前会做"clean"处理,即得到clean_data.然而,这些clean_data是否真的 ...
- Next generation sequencing (NGS)二代测序数据预处理与分析
二代测序原理: 1.DNA待测文库构建. 超声波把DNA打断成小片段,一般200--500bp,两端加上不同的接头2.Flowcell.一个flowcell,8个channel,很多接头3.桥式PCR ...
- 单细胞转录组测序数据的可变剪接(alternative splicing)分析方法总结
可变剪接(alternative splicing),在真核生物中是一种非常基本的生物学事件.即基因转录后,先产生初始RNA或称作RNA前体,然后再通过可变剪接方式,选择性的把不同的外显子进行重连,从 ...
- 基于单细胞测序数据构建细胞状态转换轨迹(cell trajectory)方法总结
细胞状态转换轨迹构建示意图(Trapnell et al. Nature Biotechnology, 2014) 在各种生物系统中,细胞都会展现出一系列的不同状态(如基因表达的动态变化等),这些状态 ...
- seqtk抽取测序数据
做数据比较的时候,由于同一个样本测序数据量不一致,需要抽取数据,控制数据量基本一致. 自己写脚本速度较慢,后面发现一个不错的工具:seqtk 原始数据抽取 如果只控制原始数据量一致,过滤低质量数据后直 ...
- 四种不同的SNP calling算法call低碱基覆盖度测序数据时,SNVs数量的比较(Comparing a few SNP calling algorithms using low-coverage sequencing data)
摘要:如果不设置任何过滤标准的话,SOAPsnp会call出更多的SNVs:AtlasSNP2算法比较严格,因此call出来的SNVs数量是最少的,GATK 和 SAMtools call出来的数量位 ...
随机推荐
- Vue之添加全局变量
定义全局变量 原理: 设置一个专用的的全局变量模块文件,模块里面定义一些变量初始状态,用export default 暴露出去,在main.js里面使用Vue.prototype挂载到vue实例上面或 ...
- github隐藏文件&删除文件
一.隐藏文件不提交至github 例如:需隐藏node_modules文件夹 1.找到.gitignore文件,一般这个是隐藏文件,需要显示隐藏文件 2.编辑.gitignore文件,加入下面这一句话 ...
- Sed练习
sed:编辑器 sed:Stream EDitor,行编辑器 用法: sed [opthon]... ‘script’ inputfile.. scritp:‘地址命令’ 常用选项: ...
- mysql权限参考
mysql日常管理和应用工作中,大家经常会涉及到授权问题,下面,我们就列举下权限相关的参考. 1.管理权限(Administrative Privileges) Privilege Name ...
- Oracle单机Rman笔记[3]---RMAN脱机备份及命令基础介绍
A.NOARCHIVELOG模式下的物理备份 1.完全关闭数据库 2.备份所有的数据库文件.控制文件.联机重做日志 3.重新启动数据库 B.RMAN的体系结构概述 重新构建控制文件: 将控制文件备份为 ...
- 让input不可编辑
有时候,我们希望表单中的文本框是只读的,让用户不能修改其中的信息,如使<input type="text" name="input1" value=&qu ...
- Daily record-October
October 11. Hope is a good thing, maybe the best of things, and no good thing ever dies. 希望是美好的,也许是人 ...
- learning makefile string example
- 路由导航之第一个子模块(HomeModule)
git clone git@github.com:len007/my-angular2-app.git my-angular2-app 开始 一个URL = 一个页面 = 一个Component. 我 ...
- flask 第七章 简陋版智能玩具 +MongoDB初识和基本操作
1.简陋版web智能玩具 FAQ.py文件 import os from aip import AipSpeech, AipNlp from uuid import uuid4 "" ...