单细胞测序技术(single cell sequencing)
|
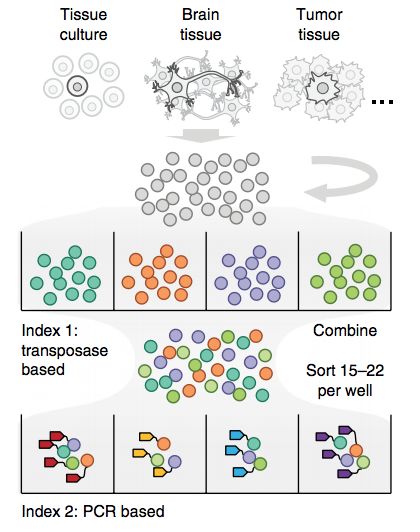
前言 单细胞生物学最近几年是非常热门的研究方向。在这一领域中,最前沿的则是单细胞测序技术。传统测序方法一次处理成千上万个细胞,得到的变异水平也是成千上万个细胞的平均后水平。但是,就如同世界上没有完全相同的两片树叶一样,没有两个细胞是完全相同的。所以,单细胞测序对于研究单个细胞就显得至关重要。 单细胞测序可以揭示出每个细胞独特的微妙变化,甚至可以揭示全新的细胞类型。单细胞测序技术可谓是科技发展史上的一大创举,它极大地推进了基因组学领域,使不同细胞类型得以精细区分,使得科学家们在单细胞水平进行分子机制研究成为可能。 随着高通量RNA测序技术的发展,2009年,开发了第一个单细胞转录组测序技术。到了2011年Nicholas等人开发了单细胞基因组测序技术。2013年又开发出了单细胞全基因组DNA甲基化检测技术。随后,科学家在细胞分选技术、核酸扩增技术、信噪比提高方面等进行不断优化和改进,也进一步开创了单细胞Hi-C、单细胞ChIP-seq、单细胞ATAC-seq技术等。 2017年单细胞测序在技术水平和应用层面上都更进一步发展,本周我们汇总了2017年单细胞测序技术层面的部分重要进展。 更多的信息请您继续关注一呼百诺。 SCI-seq 2017年3月,美国俄勒冈的研究人员在Nature Methods上发表“Sequencing thousands of single-cell genomes with combinatorial indexing (doi:10.1038/nmeth.4154) ”文章,开发出一种SCI-seq (single-cell combinatorial indexed sequencing,单细胞组合标记测序技术),多次对细胞进行条形码编码标记后对它们进行测序,可以同时构建上千个单细胞文库,检测体细胞拷贝数的变异。这项技术极大的缩减了文库构建的成本,增加了检测细胞的数量,这对于体细胞变异的检测,尤其是在肿瘤进化过程中对细胞亚克隆变异研究具有重要价值。研究人员从培养的细胞系、灵长类额叶皮层组织和两个人类胰腺癌中构建了大约17000个单细胞基因组文库,这大约比利用常规方法能够构建出的基因组文库大小高出两个数量级。
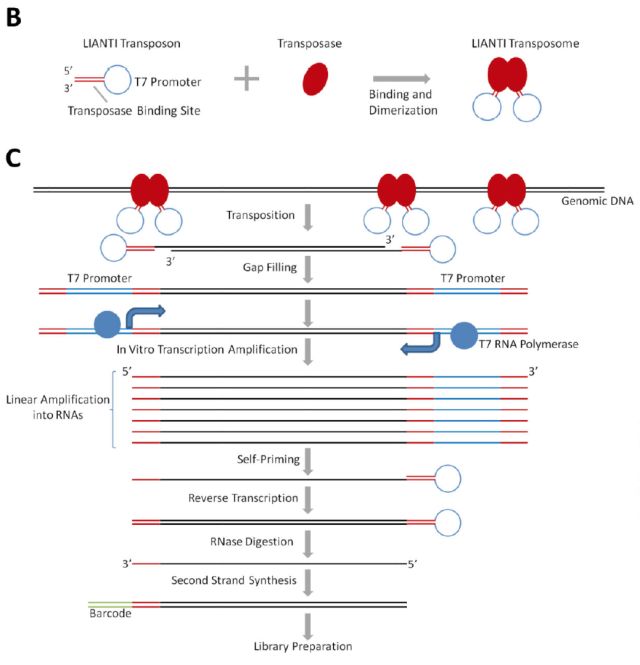
LIANTI 2017年4月北京大学谢晓亮教授团队在Science上发表“Single-Cell Whole Genome Analyses by Linear Amplification via Transposon Insertion (LIANTI) (doi: 10.1126/science.aak9787)”文章,开发一种新型的单细胞全基因组线性扩增的方法—LIANTI(Linear Amplification with Transposon Insertion)用于单细胞全基因组测序。 LIANTI法通过转座子插入进行线性放大。基因组被含有T7启动子的Tn5转座子随机片段化 ,T7启动子允许线性扩增。LIANTI法测量拷贝数的空间分辨率提高了3个数量级(能在千碱基分辨率进行微小CNV检测,基因组覆盖率可达到97%),能直接观察从细胞到细胞不同的随机发生的DNA复制起始位点。他们用新方法检测了被紫外线照射过的单个人类细胞的单核苷酸突变,结果明显优于目前已知的其他方法。这意味LIANTI能更有效、精准地检测出更多疾病突变。
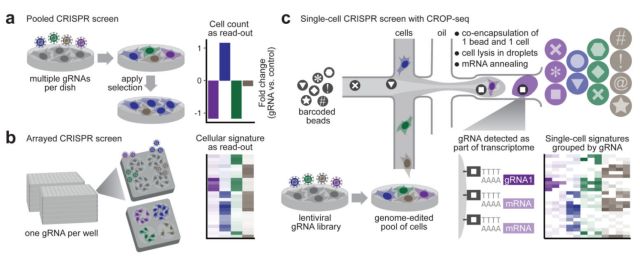
CROP-seq 2017年7月,奥地利研究人员在Nature Methods上发表“Pooled CRISPR screening with single-cell transcriptome readout (doi:10.1038/ nmeth.4227)”文章,创造性地将CRISPR-Cas9和单细胞RNA测序两种极有前景的基因组学领域结合在一起,发明了这种被称作CROP-seq的单细胞测序方法 (CRISPR droplet sequencing, CRISPR液滴测序), 能大规模完成前所未有的高通量基因调控的分析。 团队首先结合了CRISPR集中筛选和按序筛选的优势,构建出一种能够帮助研究人员看到单细胞测序实验的CRISPR gRNAs的病毒载体, 再结合最新的用于单细胞RNA测序的液滴方法足以高通量地分析单个细胞中上千种基因组编辑事件的影响。随着单细胞测序成本的下降,这种方法能够产生首批人类基因组上2.3万个基因中每个基因的调节影响的综合图谱。
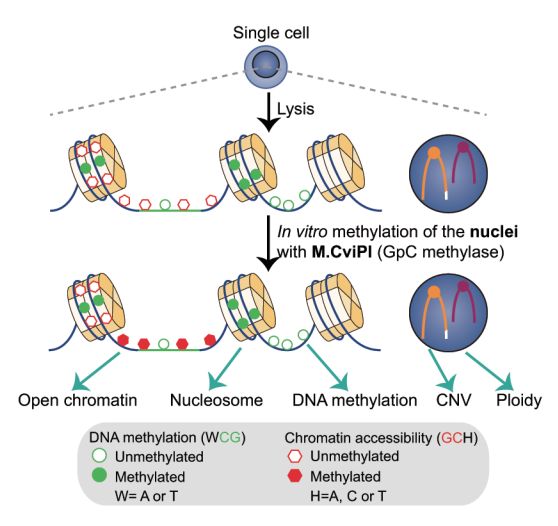
scCOOL-seq 2017年8月,北京大学汤富酬教授团队在Cell Research 上发表 “Single-cell multi-omics sequencing of mouse early embryos and embryonic stem cells (doi: 10.1038/cr.2017.82)”文章,开发了一种单细胞多重测序技术 (scCOOL-seq),可以对一个单细胞同时分析染色质状态/核小体定位、DNA甲基化、基因组拷贝数变异和染色体倍性倍性。并采用这一技术在单细胞分辨率上系统、深入地解析了小鼠着床前胚胎发育过程中表观基因组重编程的关键特征,以及染色质状态与DNA甲基化之间的互动关系。这是第一次在单细胞内对同一个单细胞进行多达5个层面的基因组和表观基因组特征的分析,突出了DNA甲基化和染色质状态的不同模式和功能。
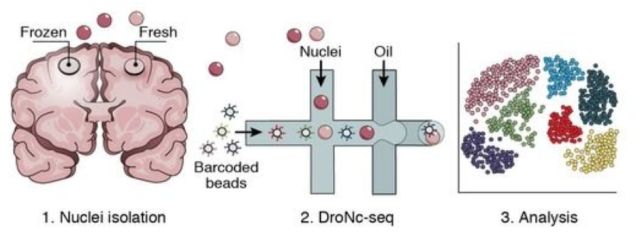
考虑到单细胞表观基因组测序技术的相对低的覆盖率,最理想的状态是明确地区分染色质状态,包括闭合状态和未检出的状态。scCOOL-seq这一新技术是可以实现这一点的单细胞染色质测序方法。此外,scCOOL-seq可以同时检测同一个体细胞中染色质状态、核小体定位、内源DNA甲基化、CNV倍性,这将大大增强单个细胞内不同遗传和表观遗传层之间的复杂关系的分析能力,为今后人们继续研究哺乳动物早期胚胎细胞全能性和多能性的开启奠定了基础,同时为体细胞克隆效率的提高以及早期胚胎发育异常的诊断与治疗提供了新思路。 DroNC-seq 10月,Broad研究所张锋教授团队在Nature Methods上发表题为“Massively Parallel Single-Nucleus RNA-Seq with DroNc-Seq (doi: 10.1038/nmeth.4407)” 的文章,提出了一个新的测序技术:DroNc-Seq单细胞表达谱分析技术。该技术融合了sNuc-Seq技术与微流控技术的单细胞核RNA测序方法,可以在结构复杂的组织中更有效地分析大量细胞或细胞核中的RNA,开启单细胞核测序的规模化时代。
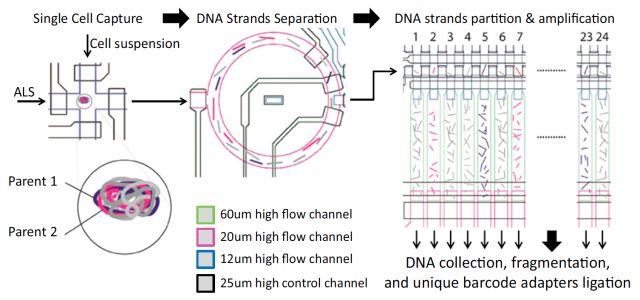
在利用单细胞技术来研究大脑等复杂组织的基因表达时,研究人员总会觉得困难重重,因为复杂组织不易分离,难以辨别,分离细胞的过程会经常破坏目的神经元和RNA。sNuc-seq则利用单个提取的细胞核作为起始材料,通过观察细胞核去理解不同的细胞类型和动态过程,如神经的形成。但sNuc-Seq是一种低通量的技术,其应用规模受到了限制。为扩大研究的应用规模,实现一次可研究数千个细胞核的高效测序,研究团队将目光转向了Drop-Seq。它将单细胞与带有DNA条码的微珠一起包裹在微滴中,大大加速表达谱分析实验,同时降低成本。最终,他们开发出了融合两种技术的DroNc-Seq方法。为检验方法的准确性与效率,研究人员利用DroNc-Seq对小鼠细胞系和脑组织进行了分析,并与Drop-Seq、sNuc-Seq以及其他较低通量的单细胞RNA-Seq方法进行比较。结果显示DroNc-Seq表现出了灵敏、高效且无偏向的细胞分类能力。该技术很可能用于人体细胞图谱计划项目。 SISSOR 11月加州大学圣地亚哥分校张鹍教授团队在PNAS上发表题为“Ultra-accurate Genome Sequencing and Haplotyping of Single Human Cells (doi: 10.1073/pnas. 1707609114)”的文章。开发了一个名为“SISSOR” (微流体反应器法单链测序)的方法,用来进行准确的单细胞基因组测序和单倍体分型。 正常人类单个细胞DNA含量为6.6pg,相对于测序所需的ug级相当微量,并且有些DNA是独一无二的,因此单细胞测序的关键一环是全基因组扩增(WGA),常见的方法如,MDA、MALBAC等需要用DNA聚合酶对细胞基因组做大量的体外扩增,然后构建文库进行相对读长较短的高通量测序。这些方法有两个明显的缺点:1,聚合酶的错误扩增可以在基因组上产生成千上万的假阳性。2,短的序列读长几乎不包含任何单倍体类型信息。
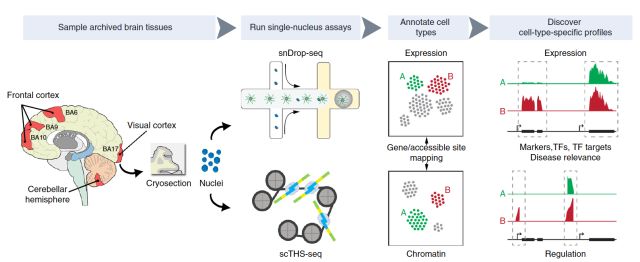
SISSOR方法是利用微流体处理器将单个细胞染色体DNA的正负双链进行分离,并将百万碱基大小的DNA片段随机分割成大量的纳升级的组分,用于扩增和构建测序文库,从而实现对同源染色体的互补双链分别进行独立的测序。这样就能够进行Long-range单倍体的组装,并且还能利用冗余和单倍体型信息纠正测序错误。结果显示,该方法的纠错能力可以将单细胞测序错误率降低至10-8,并且单倍体片段平均可组装长度为500 kb,contig达到N50大于7 Mb。该方法能够获得精准的单细胞基因组序列以及单倍体信息以满足临床对基因组测序的不同需求。SISSOR是对单细胞测序技术的一次突破,将测序准确性提高了两个数量级。这项技术潜在的应用包括对患者血液中循环肿瘤细胞进行准确的检测,以及对体外受精的健康胚胎进行筛选和检测经过基因编辑的人体细胞。 snDrop-seq+scTHS-seq 张鹍教授团队本月又在Nature Biotechnology杂志上发表了“Integrative single-cell analysis of transcriptional and epigenetic states in the human adult brain (doi: 10.1038/nbt.4038)”的文章。开发了基于冰冻组织高通量单细胞核测序方法来绘制成年人脑第二代单细胞图谱,再次引爆了单细胞研究的热点。 研究人员将视线聚焦于脑组织提取的神经元和胶质细胞单细胞核,克服了样本的限制,使该方法可以应用于新鲜或冻存的脑组织,实现了冻存组织的大规模单细胞检测。另外,这项研究的一个创新之处在于结合了两种高通量单细胞技术:基于微流体的单核测序 (snDrop-seq) 和单细胞转座子超敏感位点测序 (scTHS-seq)的方法。snDrop-seq,不仅可以对成千上万个单细胞同时进行转录组分析,从多方面对细胞类型和细胞状态进行分类,还可用于评估新鲜和冻存人体组织中的特异性表达谱,有助于研究组织的功能异质性。该技术还攻克了在微滴中有效裂解核膜而不引起RNA过度降解的难题。为同时研究表观遗传特征,该团队还开发了scTHS-seq技术。scTHS-seq拥有具有体外转录扩增的线性优势和改造后的超级突变Tn5转座酶,比ATAC-seq灵敏度更高,还提高了高度细胞特异性的远端增强子(distal enhancers)的覆盖度。
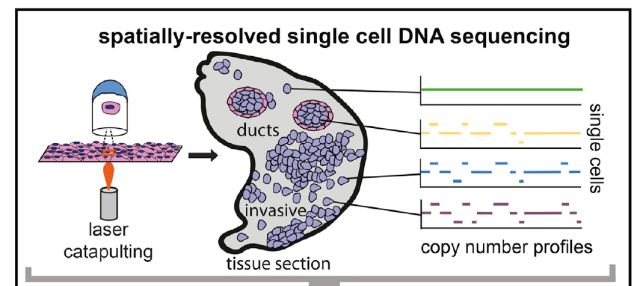
研究团队联合应用以上两种方法最终建立了这种可并行检测细胞核转录本和表观遗传特征的高通量测序平台,为综合分析冻存人体组织样本中基因表达和调节提供了途径。研究人员用此方法检测了超过60,000个来自成人大脑皮层和小脑的单细胞,发现了35种不同的神经元和神经胶质细胞亚型。此外他们还将常见的人脑遗传疾病相关的遗传位点定位到特定的细胞类型,揭示了大脑中哪些细胞类型更易受脑部疾病遗传因子的影响。这些发现有助于确定大脑中哪种类型的细胞最容易受到遗传风险因子的攻击,最终绘制一个完整的人脑细胞图谱。 TSCS 本月,美国安德森癌症中心的研究人员在Cell上发表”Multiclonal Invasion in Breast Tumors Identified by Topographic Single Cell Sequencing (doi:10.1016/ j.cell.2017.12.007)” 的研究性文章,发明了一种新的“地形”单细胞测序技术 (Topographic Single Cell Sequencing, TSCS)来研究细胞位置的空间信息,用于研究早期乳腺导管内原位癌 (DCIS)是如何发展为更具有侵袭性的导管癌症 (IDC)。 肿瘤细胞往往存在着各不相同的遗传特征,这被称为肿瘤内异质性。它们独特的细胞构成使得治疗变得更加困难。此前的单细胞DNA测序方法已经打开了探索肿瘤细胞的大门,成为了理解肿瘤内异质性的有力工具,但是这种方法能够去除关于个体肿瘤细胞在组织内精确空间定位的信息。而细胞空间数据对于了解肿瘤细胞的迁移至关重要。研究人员开发的新工具:“地形”单细胞测序 (TSCS) ,该方法提供了有关细胞位置的空间信息,能更准确地从空间上测量和描述单个肿瘤细胞的具体特征。这代表了一个技术上的里程碑,此前的技术只能采用失去了空间信息的悬浮细胞。
研究人员利用TSCS对来自10位具有DCIS和IDC的患者的1293个单细胞进行了分析。研究数据揭示了DCIS和IDC之间的直接基因组谱系,并进一步指出了入侵之前,在导管内发生的大多数突变和DNA拷贝数畸变。表明多个癌细胞克隆可以从导管共同迁移到相邻区域,形成侵入性肿瘤。 TSCS和其他类似的单细胞测序方法在开发早期癌症研究新途径方面具有巨大的潜力。希望这些研究未来能揭示为何一些恶性肿瘤前癌没有发展成恶性肿瘤,而另外一些则形成了侵入形式。 随着前不久前“人类细胞图谱”计划的开展,单细胞测序的研究热情必然持续高涨。这些单细胞测序技术的不断更新和发展无疑为构建完整细胞图谱提供了技术基础。我们可以预见,未来综合多组学的方法必将在复杂器官和组织的单细胞研究中发挥愈发强大的作用。 百诺大成拥有成熟的单细胞测序体系及后期分析流程,专业处理大量测序数据的后期深度挖掘! 作者:Elisa 校对:Shirley 编辑:李香玲 |
单细胞测序技术(single cell sequencing)的更多相关文章
- Multiclonal Invasion in Breast Tumors Identified by Topographic Single Cell Sequencing
Title: Multiclonal Invasion in Breast Tumors Identified by Topographic Single Cell Sequencing 课题的目的 ...
- 单细胞转录组测序技术(scRNA-seq)及细胞分离技术分类汇总
单细胞测序流程(http://learn.gencore.bio.nyu.edu) 在过去的十多年里,高通量测序技术被广泛应用于生物和医学的各种领域,极大促进了相关的研究和应用.其中转录组测序(RNA ...
- 单细胞参考文献 single cell
许多分析软件 : https://github.com/seandavi/awesome-single-cell#software-packages Smart-seq.CEL-seq.SCRB-se ...
- 单细胞RNA测序技术之入门指南
单细胞RNA测序技术之入门指南 [字体: 大 中 小 ] 时间:2018年09月12日 来源:生物通 编辑推荐: 在这个飞速发展的测序时代,DNA和RNA测序已经逐渐成为“实验室中的家常菜”.若要 ...
- 基于单细胞测序数据构建细胞状态转换轨迹(cell trajectory)方法总结
细胞状态转换轨迹构建示意图(Trapnell et al. Nature Biotechnology, 2014) 在各种生物系统中,细胞都会展现出一系列的不同状态(如基因表达的动态变化等),这些状态 ...
- Analysis of single cell RNA-seq data(单细胞终极课程)
业界良心啊,开源的单细胞课程. 随便看了几章,课程写得非常用心,非常适合新手. 课程地址:Analysis of single cell RNA-seq data 源码地址:hemberg-lab/s ...
- 单细胞测序|单细胞基因组|单细胞转录组|Gene editing|
单细胞测序 单细胞基因组学 测量理由是单细胞的时间空间特异性. Gene expression&co-expression 比较正常cell与疾病cell,正常organ与疾病organ,看出 ...
- Advances in Single Cell Genomics to Study Brain Cell Types | 会议概览
单细胞在脑科学方面的应用 Session 1: Deciphering the Cellular Landscape of the Brain Using Single Cell Transcript ...
- CAR-T|Single cell plan|Extracellular RNA|
生物医疗大数据 安吉丽娜朱莉发现抑癌基因事件,BRCA突变与乳腺癌关联. 个体化测序商品23 and me 多组学数据研究:eg:太空和地球双胞胎发现生化指标差不多. 研究模式和工业模式相结合. 研究 ...
随机推荐
- 历届试题 小数第n位-(同余公式+快速幂)
问题描述 我们知道,整数做除法时,有时得到有限小数,有时得到无限循环小数. 如果我们把有限小数的末尾加上无限多个0,它们就有了统一的形式. 本题的任务是:在上面的约定下,求整数除法小数点后的第n位开始 ...
- Linux简介及Linux学习路线图
一.Linux 为何物 Linux 就是一个操作系统,就像你多少已经了解的 Windows(xp,7,8)和 Max OS ,至于操作系统是什么,就不用过多解释了,如果你学习过前面的入门课程,应该会有 ...
- SpringMVC Shiro与filterChainDefinitions
SpringMVC整合Shiro,Shiro是一个强大易用的Java安全框架,提供了认证.授权.加密和会话管理等功能. 第一步:配置web.xml <!-- 配置Shiro过滤器,先让Shiro ...
- async.series
[async.series] series适用于顺序执行异步且前后无关联的调用.对于顺序执行异步且前后有叛逆的调用,则需要使用waterfall. If any functions in the se ...
- html的textarea默认文案实现换行
问题:textarea默认文案,想使用换行展示 但是使用/r/n</br>之类的都无效 解决方法: 使用下面字符实现换行 <textarea >第一行内容 第二行内容& ...
- 常用的jquerymobil 站点
http://www.jqmapi.com/api1.2/ Jquery Mobile 中文API站 https://codiqa.com/demo jquerymobil UI编辑器 https ...
- java并发:jdk1.8中ConcurrentHashMap源码浅析
ConcurrentHashMap是线程安全的.可以在多线程中对ConcurrentHashMap进行操作. 在jdk1.7中,使用的是锁分段技术Segment.数据结构是数组+链表. 对比jdk1. ...
- feign的hystrix不起作用.
在springCloud中使用feign内嵌的断路器hystrix时.feign中的hystrix不起作用.这可能是由于springCloud的版本原因造成的.需要在application.prope ...
- Django model 中的 class Meta 详解
Django model 中的 class Meta 详解 通过一个内嵌类 "class Meta" 给你的 model 定义元数据, 类似下面这样: class Foo(mode ...
- 顺时针打印矩阵(python)
题目描述 输入一个矩阵,按照从外向里以顺时针的顺序依次打印出每一个数字,例如,如果输入如下4 X 4矩阵: 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 则依次打印出数 ...