Genome Sequencing of MuseumSpecimens Reveals Rapid Changes in the Genetic Composition of Honey Bees in California
文章地址:https://academic.oup.com/gbe/article/10/2/458/4810442#supplementary-data
Abstract
在自然生态系统和管理生态系统中,Apis mellifera都是极具影响力的传粉者。在北美,这一物种已经从欧洲和非洲的各种不同来源的种群中被多次引入。从那时起,野生动物种群已经扩展到许多不同的环境,跨越了它们广泛的引进范围。在这里,我们使用了历史博物馆标本和来自美国加利福尼亚州的新收集的现代种群进行了全基因组测序,来分析群体统计学和选择对过去105年的时间里对引进种群的影响。我们发现,北加州和南加州的群体都表现出明显的基因变化,但变化的方式不同。自20世纪60年代以来,在北部种群中,蜜蜂经历了从西部到北部祖先的重大转变,而在过去20年里,非洲化的基因组向南部的渗入(introgression)则占主导地位。此外,我们还确定了一个在较长时间内变化相对较小的孤岛群体。对不同种群和时间点的精细比较也揭示了不同频率的单核苷酸多态性,突出了一些基因,这些基因可能对这些引入种群最近的适应性很重要。
Introduction
西方蜜蜂,Apis mellifera,是世界上最重要的受管理授粉者(Klein et al. 2007),也是一个标志性的家庭花园访客。自旧石器时代以来,它在欧洲、非洲和西亚的整个本土范围内都被用作蜂蜜和蜡的来源,在蜂箱中饲养蜜蜂的做法可能出现在公元前3000年至5000年的埃及。虽然在蜜蜂的自然范围内养蜂人主要依靠当地蜜蜂种群的使用,但在自然范围外养蜂人依靠各种引进的、遗传上不同的种群。这些群体现在是农业作物最重要的传粉者(Klein et al. 2007),这些群体内部和之间的遗传变异在长期存在的时空变化对农业产业至关重要。
Apis mellifera最早在1622年,被法国和英国殖民者带入美国。这些种群可能属于发生在整个西欧的M系。Apis mellifera是在美国东部的蜂巢到达SanJose市的时候,首次被引入加利福尼亚。随后,在1859年,A. mellifera蜜蜂C谱系(Ruttner 1988)被进口到北美,并很快被运到加州。尽管M系和C系在欧洲的地理位置上非常接近,但它们代表了从非洲到欧洲的两种不同的、遗传学上截然不同的辐射。然而,这两个主要的谱系自由繁殖,有admixture证据证明中欧地带的contact zone。这两种谱系是加州现代管理的蜜蜂种群的主要来源。
在加州除了大量的人工蜂群,还有大量的野生蜂群。野生蜜蜂最终来自于各种人工管理的种群,并随着成年蜜蜂的定期逃逸和建立新的野生群体而不断得到补充。虽然被管理的人工和野生的蜜蜂有共同的基因结构,但它们经历的选择压力非常不同。受管理的蜜蜂种群在获得理想性状方面承受着显著的压力,包括蜂蜜生产、病原抗性等。与此同时,由于养蜂人提供充足的食粮、住所和药物来对抗常见的病原体和寄生虫,受管理的蜜蜂不受其他选择压力的影响。而野生蜜蜂与受管理的蜜蜂相比,会经历不同的选择压力,因此,它们可能会对适应当地环境的过程提供一些洞见。这些种群甚至可以作为未来管理种群的遗传库(genetic reservoir),野生蜜蜂也为农作物授粉做出了贡献,尽管这种贡献的数量还不清楚。
1956年,来自热带非洲的 A. mellifera scutellata(lineage A)被引进巴西,它随后向加州的扩张为野生加州种群提供了另一种不同的遗传变异来源。这些种群迅速蔓延,迅速改变了美国南部野生种群的遗传面貌,尽管它们还没有完全取代来自欧洲的野生种群。非洲蜜蜂于1994年在南加州首次被记录在案。从那时起,这些种群向北迁移,2015年的一项研究发现,来自非洲的等位基因只在出现在萨克拉门托南40公里处。
在美国大陆,有近一半的成年蜂群(2016年1月为1140万只)季节性地生活在加州。这使得养蜂者和农业产业对有关加利福尼亚野生和管理蜜蜂种群基因组成变化的信息特别有兴趣。A. mellifera群 在过去的几十年里,在加州经历了一系列的选择压力。首先,人们担心的是非洲化的蜜蜂及这些蜜蜂与人工的蜂群杂交的潜力,产生具有unfavorable特征的后代(攻击性行为,降低蜂蜜产量)。其次,在美国,人工管理锋群的死亡率在1944年和2008年这期间有所上升。2016年,美国农业部报告说,从1月到3月,加利福尼亚的蜂群减少了15%。这些损失被认为是由许多因素造成的,包括杀虫剂,疾病和寄生虫,这些因素目前威胁到成年蜜蜂的数量。然而,有迹象表明,基因多样化的蜂箱可能对疾病更有抵抗力。
在此之前的几项研究中,曾试图量化不同A. mellifera lineages对加利福尼亚和美国的总体种群的贡献,但、要么使用线粒体标记,要么使用少许基因来评估种群结构。在此,我们提出了一个基于全基因组的研究来评估过去105年(1910-2015)引入的欧洲和非洲蜜蜂种群(野生和人工)对加利福尼亚蜜蜂的贡献。我们对祖先和基因如何随着时间、地域和种群的变化而变化的分析,为适应性基因位点和基因分化提供了深刻的见解,而且是了解生态相关特性和局部适应的第一步。为了进行我们的分析,我们使用了来自整个州的配对的历史和现代samples,跨越了蜜蜂已经在加州存在65%的时间。我们假设,在空间和时间维度上,不同的欧洲和非洲祖先谱系对加利福尼亚群体的遗传特征的贡献发生了重大变化。特别的,我们希望看到随着时间的推移,African lineages对加利福尼亚群体的贡献有所增加,包括鉴定admixed individuals,非洲蜜蜂平行的到来和传播(arrival and spread).我们进一步假设了在瓦螨爆发后的几年里,由于野生种群遭受了巨大的损失,以及最近从人工管理种群中获得的野生种群的预期比例增加,北加利福尼亚基因多样性会减少。最后,我们希望找出可能与加州不同生态区域的局部迁移有关的候选基因。
Materials and Methods
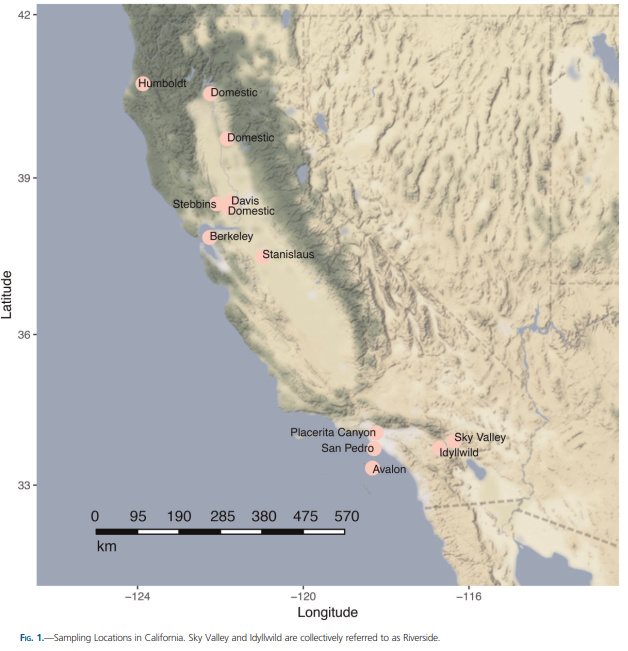
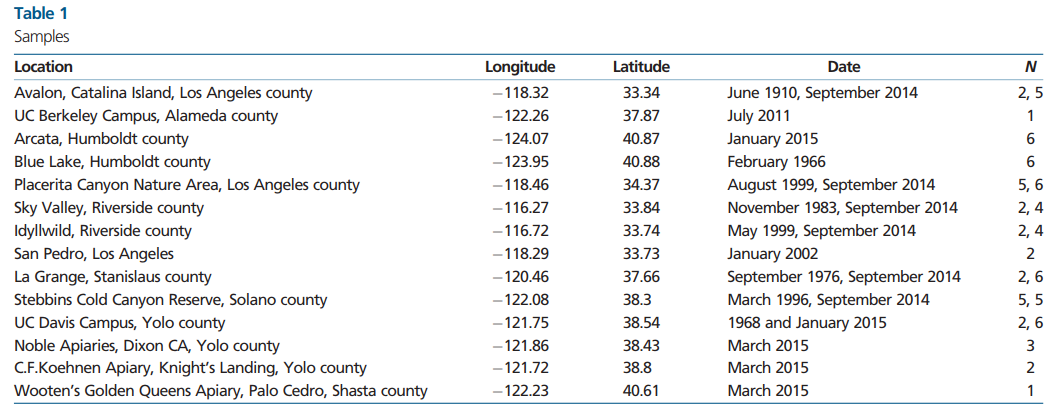
我们收集了29个1910年至2011年间的加利福尼亚博物馆收藏的A. mellifera标本,(图1和表1)。:此外,我们还从两个公开可用的数据集中包含了一组A . mellifera个体。
我们使用了之前提取的SNP数据集(Cridland et al. 2017)来推断从加州引进的 A. mellifera种群的祖先模式。这组样本来自非洲(47个个体)、西欧(39个个体)和东欧(29人)、中东(10个体)和10日本个体。用于博物馆和现代标本(modern samples)的DNA提取和文库所有步骤都使用了过滤器提示。博物馆的标本是单独处理的,与现代标本是分开处理的。准备工作是相似的。在使用前,用漂白剂清洗台式、移液管和外用镊子,并用蒸馏水冲洗干净。博物馆的标本是单独处理的,与现代标本是分开处理的。对于用于现代标本,DNA提取使用DNeasy Blood and Tissue kit (Qiagen)遵循标准方案,博物馆标本则用减少的洗脱体积(130毫升)。采用真空离心浓缩器对量子位读数小于2.5 ng/ml的样品进行DNA浓缩。
全基因组文库制备完成Nextera DNA文库制备试剂盒(Illumina)遵循低丛索引池的标准协议。 Illumina HiSeq2000产生 100 bp single-end reads 在文森特J加州大学伯克利分校科茨基因组测序实验室。序列与A. mellifera参考基因组版本4.5(可从beebase.org获得)进行比对,使用带有非常敏感的局部比对参数的Bowtie2。变异(variation)的覆盖(coverage )范围很大程度上是由于从保存的博物馆标本中提取高质量DNA进行测序的困难,而我们最古老的序列,尤其是1910年的序列,平均覆盖范围最低(补充表1)。较老的样本也更有可能丢失数据,尽管除了一个样本外,所有样本都至少包含了一半的snp。

Supplementary Table 1: FST Differences between Californian Populations
SNP Sets
我们生成了两组snp来分析加利福尼亚的样本。第一个snp是以前生成的一组在非洲和欧洲本土蜜蜂中发现的snp(Cridland et al. 2017),并用于所有祖先分析。我们使用samtools/vcftools为每一个单独的个体生成SNP调用,需要的质量值才能包含读取。每个个体最低 需要7×coverage进行genotype calls,而且我们需要最低2×coverage进行heterozygote calls。第二组是从2014年至2015年采集的蜜蜂样本,用于调查加州群体之间的差异。因为大多历史样本覆盖水平较低,因此更少的有基因型信息位点,我们使用了2014个样本(41野生+6人工)来确定下游一组SNP组分析。为了在数据集中包含SNP,我们要求能够在2014/2015年的40/47个样本中调用一个基因型。然后我们在所有样本中的这些位置使用了与上面相同的一组覆盖率要求进行 genotype calls 。共鉴定了3,890,276个snp,用于下游分析。
Ancestry Analyses
ADMIXTURE软件,对于完整的欧洲、中东、非洲和加利福尼亚的个体,K种群值在2到6之间,以检验种群在不同的假设值上分裂(population splits at different assumed values)。我们最初对包括O组个体在内的数据进行了分析,但这些个体从未被ADMIXTURE从C组个体中分离出来。在删除O组的情况下运行分析,产生了关于Californian个体一致性的结果。因此,我们的结论是,要么是O组没有对加州的群体作出贡献,要么是我们无法将来自O组的贡献与来自C组的贡献分离开来。我们发现个体来自河滨县(Sky)的两个地点谷地和田园野地)在每次分析中都非常相似,我们将这两个地点合并成一个种群进行下游分析。
我们使用Dadi对2014年收集到的加州种群及来自非洲、西欧和东欧的种群对进行了FST计算。为了检验个体和主要群体之间的关联模式,我们使用R包ecodist version 1.2.2进行了非参数多维尺度分析(nMDS),并计算了每个分析的应力值( stress value)和R2值。nMDS分析通过在多维空间中定位个体来表示个体之间的相似性。我们在R中运行了adonis函数(版本2.3-4)包,以确定主要分组对距离矩阵的影响。
我们用ADMIXTOOLS对admixture进行了正式的试验。我们计算每一潜在目标群体的所有源群体对的f3,以确定有admixture证据的群体,即f3为负值。对于确定的负f3统计量的测试,我们计算了admixture比例的上限和下限。然后我们保存了所有发现admixture证据的结果( f3 <-0.01),z分数离均值至少有四个标准差远.F4比值预测是在通过我们的过滤器估计混合种群中祖先比例的同一组f3统计数据上进行的。
Geographic Differentiation
我们计算了每一地点之间的非洲,西欧、东欧、加州2014种群对的FST。
Genome Sequencing of MuseumSpecimens Reveals Rapid Changes in the Genetic Composition of Honey Bees in California的更多相关文章
- 全基因组测序 Whole Genome Sequencing
全基因组测序 Whole Genome Sequencing 全基因组测序(Whole Genome Sequencing,WGS)是利用高通量测序平台对一种生物的基因组中的全部基因进行测序,测定其 ...
- adaptation|domestication|genome evolution|convergent evolution|whole-genome shotgun sequencing|IHGSC
Dissecting evolution and disease using comparative vertebrate genomics-online 因为基因组不是独一无二的,同时人类基因组可以 ...
- GATK使用说明-GRCh38(Genome Reference Consortium)(二)
Reference Genome Components 1. GRCh38 is special because it has alternate contigs that represent pop ...
- 测序深度和覆盖度(Sequencing depth and coverage)
总是跑数据,却对数据一无所知,这说不过去吧. 看几篇文章吧 Sequencing depth and coverage: key considerations in genomic analyses( ...
- 单细胞测序技术(single cell sequencing)
单细胞测序技术(single cell sequencing) 2018-03-02 11:02 来源: 一呼百诺 点击次数:6587关键词: 前言 单细胞生物学最近几年是非常热门的研究方向 ...
- METAGENOMIC SEQUENCING ANALYSIS WORKFLOW
Metagenomics is defined as the study of the metagenome, which is total genomic DNA from environmenta ...
- The top 100 papers Nature explores the most-cited research of all time.
The top 100 papers Nature explores the most-cited research of all time. The discovery of high-temper ...
- 2016-6-15-de novo文献阅读
准备读四篇denovo的文献: Nature Biotechnology(2015) - Sequencing of allotetraploid cotton (Gossypium hirsutum ...
- 【bioinfo】生物信息学——代码遇见生物学的地方
注:从进入生信领域到现在,已经过去快8年了.生物信息学包含了我最喜欢的三门学科:生物学.计算机科学和数学.但是如果突然问起,什么是生物信息学,我还是无法给出一个让自己满意的答案.于是便有了这篇博客. ...
随机推荐
- LeetCode 26:删除排序数组中的重复项 Remove Duplicates from Sorted Array
给定一个排序数组,你需要在原地删除重复出现的元素,使得每个元素只出现一次,返回移除后数组的新长度. 不要使用额外的数组空间,你必须在原地修改输入数组并在使用 O(1) 额外空间的条件下完成. Give ...
- 一个简单的利用 WebClient 异步下载的示例(三)
继续上一篇 一个简单的利用 WebClient 异步下载的示例(二) 后,继续优化它. 1. 直接贴代码了: DownloadEntry: public class DownloadEntry { p ...
- JVM的监控工具之jstat
参考博客:https://www.cnblogs.com/lxcmyf/p/9878293.html jstat(JVMStatisticsMonitoringTool)是用于监视虚拟机各种运行状态信 ...
- kali渗透综合靶机(六)--FristiLeaks靶机
kali渗透综合靶机(六)--FristiLeaks靶机 靶机地址下载:https://download.vulnhub.com/fristileaks/FristiLeaks_1.3.ova 一.主 ...
- duba网址对firefox快捷方式的劫持
直接删除 “驱动精灵” 即可. 等我 二进制安全 学好了,一定开发一种病毒专干这种劫持的,煞笔软件.
- 搭建 Frp 来远程内网 Windows 和 Linux 机子
魏刘宏 2019 年 5 月 19 日 一.使用一键脚本搭建服务端 Frp 这个内网穿透项目的官方地址为 https://github.com/fatedier/frp ,不过我们今天搭建服务端时不直 ...
- C# - VS2019 WinFrm程序调用ZXing.NET实现条码、二维码和带有Logo的二维码的识别
前言 C# WinFrm程序调用ZXing.NET实现条码.二维码和带有Logo的二维码的识别. ZXing.NET导入 GitHub开源库 ZXing.NET开源库githib下载地址:https: ...
- git和小乌龟在windows下安装
一:所需软件 (1):git 下载地址:https://git-scm.com/download (2):TortoiseGit 下载地址:https://tortoisegit.org/downlo ...
- Scrapy-Splash简介及验证码的处理(一)
目录 一:Splash简介与准备 二:验证码的识别(1) 在之前的博客中,我们学习了selenium的用法,它是一个动态抓取页面的方法,但是,动态抓取页面还有其他的方法,这里介绍Splash方法, ...
- 将多个sass文件合并到一个文件中
将多个sass文件合并到一个文件中 应用场景:制作angular npm包的时候,定义的一些全局样式,自定义主题色这类的情况下,多个scss文件会要合并成一个文件并写到dist文件里,发布到仓库中. ...