10X Genomics vs. PacBio
10X Genomics已经广泛应用于单细胞测序、组装领域,现在也是火的不行。
10X Genomics原理
通过将来自相同DNA片段(10-100kb)的reads加上相同的barcode,然后在illumina平台上进行测序,从而实现长片段的测序。其基本原理是同一长片段的reads会具有同样的标签,称为linked-reads,利用这些barcode的信息,可将短reads拼接为长reads。这样的linked-reads可进行结构变异检测及单倍型定相的分析。
总结:10X Genomics发明了一种独特的barcode建库技术,能将同一种barcode的reads连成linked reads从而提高了reads的长度。但10X Genomics的测序之本仍然是illumina,GC偏向性是硬伤。同时barcode技术有一定的环境限制。
附:未来基因组组装的标配为“PacBio纯三代组装contig“+”光学图谱进行纠错与super scaffold组装“+”遗传图谱或HiC进行染色体组装”。
DNA分析流程

RNA分析流程

转自:菲沙基因
2015年测序行业杀出一匹黑马,10X Genomics公司,他们开发了一个巧妙的方法,能使Illumina测序仪产生长片段数据。What?!100-300bp变成10-100kb,巨大的片段长度提升一时间吸引了无数人的眼球。提到长片段测序,永远都绕不开 PacBio。下面我们来一起看看,10X Genomics是如何挑战PacBio,而PacBio又是如何应对10X Genomics的“来势汹汹”。
1巧妙的平台互补
GEM反应体系中的DNA模板打断后与Barcode结合形成适合建库的小片段,配合Illumina测序得到原始数据。GemCode平台配套的数据分析软件将Barcode标记能力与短读取数据相结合,产生了独特的一类数据类型:Linked-Reads,利用Barcode标记信息将同一模版DNA来源的序列信息进行拼接,从而获得大片段遗传信息(10-100kb)。10X Genomics官方宣传这种Linked-Reads数据让研究人员能够以前所未有的水平检测单体型和结构变异。同时,缺失、重复和重排等具有挑战性的结构变异也能够自信鉴定,从而更准确地了解基因组复杂性。Fig.1 GEMcode平台原理 10X Genomics 公司开发的GemCode平台是一套分子条形码和分析系统,由仪器、试剂盒和信息学软件组成。目前,它只与Illumina测序系统兼容。GemCode技术的核心是对1 ng的DNA进行精确分区a,形成含有1条DNA模板链和特定相同的Barcode序列的微小反应体系(GEM,Gel Bead in Emulsion),不同GEM反应体系中的Barcode不同。

2惊人的组装效果
去年PacBio公司在《Nature Methods》上发表了SMRT测序组装人类基因组的成果,选用的样本是HapMap样品NA12878[1]。而在今年5月,10X Genomics很“偶然地”选择了同样的样本,结合short-read测序和10X的linked-read测序对人类基因组进行了高质量de novo组装,并且该成果也很“偶然地”发表于《Nature Methods》上。相同的样本,相似的长片段,给人带来无限联想,看来10X Genomics已经卯足了劲准备“开大”了。
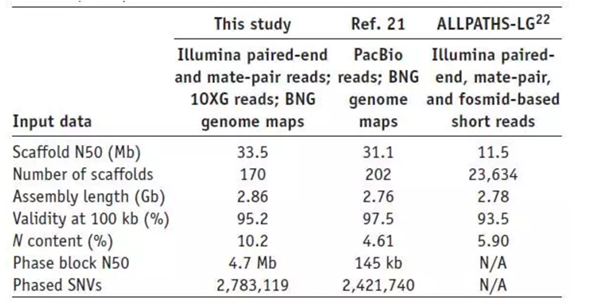
F ig.2 NA12878组装效果对比
在10X Genomics研究中,研究人员利用这种方法对人类HapMap样品NA12878进行组装和分相。最初的Illumina组装产生了超过14,000个scaffold,而N50为0.59 Mb。在混合组装后,scaffold数量降为170个,而N50大小达到33.5 Mb,相对之前提高了57倍[2]。从组装结果来看,不管是N50大小、组装长度还是scaffold数,10X Genomics的组装效果都要强于PacBio。这一局,看起来10X Genomics赢了,事实真的是这样吗,我们继续往下看。
3长片段优劣之争
一种新技术的出现,往往会伴随着争议。10X Genomics到底有没有它宣传的那么好,我们结合技术原理和文章来仔细分析一下。
1) 长片段的产生
简单来说,10X Genomics获得长片段的方法基于DNA精确分区和Barcode技术。将100kb长片段模板和Barcode混合组成一个GEM,打断后该体系中每个短片段都带有相同Barcode。使用Illumina平台对短片段进行测序,然后使用Barcode即可将短片段拼接成长片段。这种方法对DNA要求高,长期保存的样品往往无法使用。另外,一个文库中linked-read的长片段模板不一定会全部被扩增,所以该方法也需要构建不同大小的多个文库。相对而言,PacBio的长片段直接由测序读取,无需拼接,一致准确性和覆盖度都较高。
2) GC异常区域的检测
去年10月《Nature Review Genetics》的一篇文章中总结了Illumina、10X Genomics和PacBio技术在不同GC含量区域的覆盖度分布情况[3]。结果显示,虽然10X Genomic比Illumina测序覆盖效果有所提升,但是两者整体的一致覆盖度趋势相似,都呈拱形,即对高GC或低GC区域的覆盖效果都比较差。而PacBio在这方面效果良好,基本无偏倚。从本质上看,10X Genomics只是将Illumina短片段变长,并没有彻底消除Illumina平台GC偏好的问题。归根结底,是否进行PCR扩增是影响10X Genomics和PacBio长片段覆盖度的主要因素。

Fig.3 各测序平台不同GC含量区域覆盖度比较
3) 结构变异检测
既然都能得到长片段,那么各平台在SV检测方面的能力又怎样呢,我们通过两张图来对比一下。

Fig.4 SV检测
10X Genomics今年2月在《Nature Biotechnology》发表文章,主打结构变异检测和单体型分相。左图是10X Genomics关于缺失变异的检测结果,通过数据覆盖度的趋势拟合结构变异特征,从图中可以看出Chr8 39.3Mb处出现high-confidence deletion[4]。右图是PacBio去年《Nature Methods》文章中的sv检测结果[1],粉红部分代表deletion,灰色部分代表Inversion。我只想说,PacBio太凶残了有木有,10X Genomics只给出个趋势,你偏偏要画出准确结构!
最后说两句,Illumina测序是10X Genomics的根,Illumina做不到的,10X Genomics也无能为力。PacBio虽然贵,但是贵得值,毕竟花多少钱做多大事!
4参考文献
1. Pendleton M, Sebra R, Pang A W, et al. Assembly and diploid architecture of an individual human genome via single-molecule technologies.[J]. Nature Methods, 2015, 12(8).
2. Zheng G X, Lau B T, Schnall-Levin M, et al. Haplotyping germline and cancer genomes with high-throughput linked-read sequencing.[J]. Nature Biotechnology, 2016.
3. Chaisson M J P, Wilson R K, Eichler E E. Genetic variation and the de novo assembly of human genomes[J]. Nature Reviews Genetics, 2015, 16(11):627-40.
4. Mostovoy Y, Levy-Sakin M, Lam J, et al. A hybrid approach for de novo human genome sequence assembly and phasing.[J]. Nature Methods, 2016.
10X Genomics vs. PacBio的更多相关文章
- Cell theory|Bulk RNA-seq|Cellar heterogeneity|Micromanipulation|Limiting dilution|LCM|FACS|MACS|Droplet|10X genomics|Human cell atlas|Spatially resolved transcriptomes|ST|Slide-seq|SeqFISH|MERFISH
生物信息学 Cell theory:7个要点 All known living things are made up of one or more cells. All living cells ar ...
- 10X genomics|cell base|in-vivo based|model organisms|SBI|
生命组学-药物基因组学 精准医学的内容有个人全基因组测序,移动可穿戴设备,它可以实时监测,深度学习模型预测疾病,对疾病预测做到有效.安全和可控. 药物基因组学就是研究疾病.化合物和靶点之间的关系,关键 ...
- 单细胞 RNA-seq 10X Genomics
单细胞流程跑了不少,但依旧看不懂结果,是该好好补补了. 有些人可能会误会,觉得单细胞的RNA-seq数据很好分析,跟分析常规的RNA-seq应该没什么区别.今天的这篇文章2015年3月发表在Natur ...
- 基因组Denovo组装原理、软件、策略及实施
目录 1. 组装算法 1)基于OLC算法 2)基于DBG算法 3)OLC vs DBG 2. 组装软件 3. 组装策略 4. 组装项目实施 1)测序前的准备 2) 测序样品准备 3)测序策略的选择 4 ...
- Canu Quick Start(快速使用Canu)
Canu Quick Start Canu Quick Start PBcR (老版的canu) CA Canu specializes in(专门从事) assembling PacBio or O ...
- 单细胞数据初步处理 | drop-seq | QC | 质控 | 正则化 normalization
比对 The raw Drop-seq data was processed with the standard pipeline (Drop-seq tools version 1.12 from ...
- 单细胞RNA测序技术之入门指南
单细胞RNA测序技术之入门指南 [字体: 大 中 小 ] 时间:2018年09月12日 来源:生物通 编辑推荐: 在这个飞速发展的测序时代,DNA和RNA测序已经逐渐成为“实验室中的家常菜”.若要 ...
- 文献阅读 | Molecular Architecture of the Mouse Nervous System
文章亮点: 按level来管理和分析数据,文章有不同stage,每个stage有不同subtype,这应该是一个真tree,而不只是一个进化树,文章里出现最多的进化树把所有的stage都整合了. 空间 ...
- 单细胞测序|单细胞基因组|单细胞转录组|Gene editing|
单细胞测序 单细胞基因组学 测量理由是单细胞的时间空间特异性. Gene expression&co-expression 比较正常cell与疾病cell,正常organ与疾病organ,看出 ...
随机推荐
- nginx之rewrite重写,反向代理,负载均衡
rewrite重写(伪静态): 在地址栏输入xx.com/user-xxx.html, 实际上访问的就是xxx.com/user.php?id=xxx rewrite就这么简单 附上ecshop re ...
- CodeForces - 946D Timetable (分组背包+思维)
题意 n天的课程,每天有m个时间单位.若时间i和j都有课,那么要在学校待\(j-i+1\)个时间.现在最多能翘k节课,问最少能在学校待多少时间. 分析 将一天的内容视作一个背包的组,可以预处理出该天内 ...
- 向SVN添加新项目的实践笔记
假设已经安装好SVN服务器端和客户端 1.在SVN上新建文件夹: 2.进入工程项目所在文件夹,鼠标右键菜单选择[SVN Checkout...],弹出如下对话框 3.点击第2步对话框OK按钮,弹出如下 ...
- 拉取远程仓库到本地错误The authenticity of host 'github.com (13.229.188.59)' can't be established.
1.个人在github上面创建了仓库,通过本地的git拉取远程仓库到本地报错信息如下: 这是因为Git使用SSH连接,而SSH第一次连接需要验证GitHub服务器的Key.确认GitHub的Key的指 ...
- 访问Hsql .data数据库文件
一.Hsql简介: hsql数据库是一款纯Java编写的免费数据库,许可是BSD-style的协议. 仅一个hsqldb.jar文件就包括了数据库引擎,数据库驱动,还有其他用户界面操作等内容.下载地址 ...
- 解析分布式锁之Redis实现(二)
摘要:在前文中提及了实现分布式锁目前有三种流行方案,分别为基于数据库.Redis.Zookeeper的方案,本文主要阐述基于Redis的分布式锁,分布式架构设计如今在企业中被大量的应用,而在不同的分布 ...
- bzoj1635 / P2879 [USACO07JAN]区间统计Tallest Cow
P2879 [USACO07JAN]区间统计Tallest Cow 差分 对于每个限制$(l,r)$,我们建立一个差分数组$a[i]$ 使$a[l+1]--,a[r]++$,表示$(l,r)$区间内的 ...
- troubleshooting-When importing query results in parallel, you must specify --split-by.
原因分析 -m 4 \ 导数命令中map task number=4,当-m 设置的值大于1时,split-by必须设置字段(需要是 int 类型的字段),如果不是 int类型的字段,则需要加上参数- ...
- keepalived+nginx实现HA高可用的web负载均衡
Keepalived 是一种高性能的服务器高可用或热备解决方案, Keepalived 可以用来防止服务器单点故障的发生,通过配合 Nginx 可以实现 web 前端服务的高可用.Keepalived ...
- tf.truncated_normal的用法
tf.truncated_normal(shape, mean, stddev) :shape表示生成张量的维度,mean是均值,stddev是标准差.这个函数产生正太分布,均值和标准差自己设定.这是 ...