edgeR使用学习【转载】
转自:http://yangl.net/2016/09/27/edger_usage/
1.Quick start

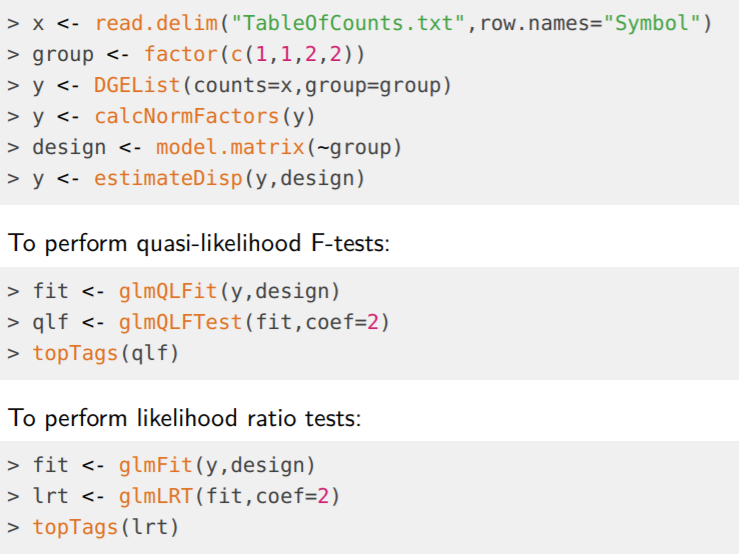
2. 利用edgeR分析RNA-seq鉴别差异表达基因:
#加载软件包
library("edgeR",verbose=0); # 1. 载入数据 读取read count数
data <- read.delim("pnas_expression.txt", row.names=1, stringsAsFactors=FALSE);
head(data); #输出
# lane1 lane2 lane3 lane4 lane5 lane6 lane8 len
# ENSG00000215696 0 0 0 0 0 0 0 330
# ENSG00000215700 0 0 0 0 0 0 0 2370
# ENSG00000215699 0 0 0 0 0 0 0 1842
# ENSG00000215784 0 0 0 0 0 0 0 2393
# ENSG00000212914 0 0 0 0 0 0 0 384
# ENSG00000212042 0 0 0 0 0 0 0 92 dim(data);
# [1] 37435 8 #2. 构建分组变量
#分为 Control组和DHT组 分别为4个和3个重复
targets <- data.frame(Lane = c(1:6,8), Treatment = c(rep("Control",4),rep("DHT",3)),
Label = c(paste("Con", 1:4, sep=""), paste("DHT", 1:3, sep=""))); targets
#输出
# Lane Treatement Label
# 1 1 Control Con1
# 2 2 Control Con2
# 3 3 Control Con3
# 4 4 Control Con4
# 5 5 DHT DHT1
# 6 6 DHT DHT2
# 7 8 DHT DHT3 #3. 创建基因表达列表 进行标准化因子计算
y <- DGEList(counts=data[,1:7], group=targets$Treatment, genes=data.frame(Length=data[,8]));
colnames(y) <- targets$Label;
dim(y);
# [1] 37435 7 #过滤表达量偏低的基因 !!!
#基因在至少3个样本中得count per million(cpm)要大于1
keep <- rowSums(cpm(y)>1) >= 3;
y <- y[keep,];
dim(y)
# [1] 16494 7
#重新计算库大小
y$samples$lib.size <- colSums(y$counts); #3. 进行标准化因子计算 默认使用TMM方法
y <- calcNormFactors(y);
y #输出
# An object of class "DGEList"
# $counts
# Con1 Con2 Con3 Con4 DHT1 DHT2 DHT3
# ENSG00000124208 478 619 628 744 483 716 240
# ENSG00000182463 27 20 27 26 48 55 24
# ENSG00000124201 180 218 293 275 373 301 88
# ENSG00000124207 76 80 85 97 80 81 37
# ENSG00000125835 132 200 200 228 280 204 52
# 16489 more rows ...
#
# $samples
# group lib.size norm.factors
# Con1 1 976847 1.0296636
# Con2 1 1154746 1.0372521
# Con3 1 1439393 1.0362662
# Con4 1 1482652 1.0378383
# DHT1 1 1820628 0.9537095
# DHT2 1 1831553 0.9525624
# DHT3 1 680798 0.9583181
#
# $genes
# [1] 2131 5453 4060 3264 945
# 16489 more rows ... #这里主要是通过图形的方式来展示实验组与对照组样本是否能明显的分开
#以及同一组内样本是否能聚的比较近 即重复样本是否具有一致性
plotMDS(y); #4. 估计离散度
y <- estimateCommonDisp(y, verbose=TRUE)
# Disp = 0.02002 , BCV = 0.1415
y <- estimateTagwiseDisp(y); plotBCV(y); #5. 通过检验来鉴别差异表达基因
et <- exactTest(y);
top <- topTags(et);
top #输出
# Comparison of groups: DHT-Control
# Length logFC logCPM PValue FDR
# ENSG00000151503 5605 5.816156 9.716866 0.000000e+00 0.000000e+00
# ENSG00000096060 4093 5.004454 9.950606 0.000000e+00 0.000000e+00
# ENSG00000166451 1556 4.683425 8.850401 2.297717e-249 1.263285e-245
# ENSG00000127954 3919 8.120955 7.216393 1.534440e-229 6.327264e-226
# ENSG00000162772 1377 3.316701 9.743797 7.975243e-216 2.630873e-212
# ENSG00000115648 2920 2.598440 11.474677 6.984860e-180 1.920138e-176
# ENSG00000116133 4286 3.244446 8.791930 1.290432e-174 3.040627e-171
# ENSG00000113594 10078 4.111120 8.055613 3.115276e-161 6.422921e-158
# ENSG00000130066 868 2.609899 9.989778 6.009018e-155 1.101253e-151
# ENSG00000116285 3076 4.201846 7.361640 6.299060e-149 1.038967e-145 #6. 定义差异表达基因与基本统计
summary(de <- decideTestsDGE(et)); # 默认选取FDR = 0.05为阈值 #输出
# [,1]
# -1 2094 #显著下调
# 0 12060 #没有显著差异
# 1 2340 #显著上调 #图形展示检验结果
detags <- rownames(y)[as.logical(de)];
plotSmear(et, de.tags=detags);
abline(h=c(-1, 1), col="blue");
//这个是分为 Control组和DHT组,检验这两组的差异表达基因。
//中间又一步是去除表达量过低的基因。
- 读取read count数
- 构建分组变量
- 创建基因表达列表 进行标准化因子计算 ,过滤表达量偏低的基因,进行标准化因子计算 默认使用TMM方法
- 估计离散度
- 通过检验来鉴别差异表达基因
- 定义差异表达基因与基本统计
edgeR使用学习【转载】的更多相关文章
- Java多线程学习(转载)
Java多线程学习(转载) 时间:2015-03-14 13:53:14 阅读:137413 评论:4 收藏:3 [点我收藏+] 转载 :http://blog ...
- Windows Services 学习(转载)
转载:http://blog.csdn.net/fakine/article/details/42107571 一.学习点滴 1.本机服务查看:services.msc /s2.服务手动安装(使用sc ...
- 【学习转载】MyBatis源码解析——日志记录
声明:转载自前辈:开心的鱼a1 一 .概述 MyBatis没有提供日志的实现类,需要接入第三方的日志组件,但第三方日志组件都有各自的Log级别,且各不相同,但MyBatis统一提供了trace.deb ...
- JVM的相关知识整理和学习--(转载)
JVM是虚拟机,也是一种规范,他遵循着冯·诺依曼体系结构的设计原理.冯·诺依曼体系结构中,指出计算机处理的数据和指令都是二进制数,采用存储程序方式不加区分的存储在同一个存储器里,并且顺序执行,指令由操 ...
- Jqgrid学习(转载)
jqGrid API 全 JQGrid是一个在jquery基础上做的一个表格控件,以ajax的方式和服务器端通信. JQGrid Demo 是一个在线的演示项目.在这里,可以知道jqgrid可以做 ...
- R中双表操作学习[转载]
转自:https://www.jianshu.com/p/a7af4f6e50c3 1.原始数据 以上是原有的一个,再生成一个新的: > gene_exp_tidy2 <- data.fr ...
- Java核心编程快速学习(转载)
http://www.cnblogs.com/wanliwang01/p/java_core.html Java核心编程部分的基础学习内容就不一一介绍了,本文的重点是JAVA中相对复杂的一些概念,主体 ...
- FPGA/SOPC学习转载
转自小時不識月http://www.cnblogs.com/yuphone/archive/2010/08/27/docs_plan.html 新网址为:http://andrewz.cn [连载计划 ...
- OpenGL入门学习(转载)
说起编程作图,大概还有很多人想起TC的#include <graphics.h>吧? 但是各位是否想过,那些画面绚丽的PC游戏是如何编写出来的?就靠TC那可怜的640*480分辨率.16色 ...
随机推荐
- C语言字节对齐问题详解
引言 考虑下面的结构体定义: typedef struct{ char c1; short s; char c2; int i; }T_FOO; 假设这个结构体的成员在内存中是紧凑排列的,且c1的起始 ...
- 【C#新特性】不用out ref同时返回多个值-元组Tuple
元组Tuple,它是一种固定成员的泛型集合 下面先看看官方的一个使用例子: 创建一个包含7个元素的Tuple数组 // Create a 7-tuple. , , , , , );// Display ...
- ANDROID – 單色漸層效果的改良 – GRADIENT SCRIMS(转)
本篇是根據 +Roman Nurik 在 2014/11/24 發佈的一篇 G+ 而來.看到他發文後,起了好奇心,就根據他提出的方法嘗試著實作,並將之排列呈現,直接從視覺上做個比較. 他在 G+ 的發 ...
- delphixe10 android操作 打电话,摄像头,定位等
XE6 不支持JStringToString.StringTojString.StrToJURI:use Androidapi.Helpers //Splash Image Delphi XE5,XE ...
- JS方法 - 字符串处理函数封装汇总 (更新中...)
一.计算一段字符串的字节长度 字符串的charCodeAt()方法, 可返回字符串固定位置的字符的Unicode编码,这个返回值是0-65535之间的整数,如果值<=255时为英文,反之为中文. ...
- Ubuntu 16.04 LAMP server tutorial with Apache 2.4, PHP 7 and MariaDB (instead of MySQL)
https://www.howtoforge.com/tutorial/install-apache-with-php-and-mysql-on-ubuntu-16-04-lamp/ This tut ...
- sencha touch Button Select(点击按钮进行选择)扩展
此扩展基于官方selectfield控件修改而来,变动并不大,使用方法类似. 代码如下: Ext.define('ux.SelectBtn', { extend: 'Ext.Button', xtyp ...
- Ico初步理解
Ico定义:是一个重要的面向对象编程的法则来削减计算机程序的耦合问题(解耦).通俗理解:把运行中程式的控制权从程式本身那里拿过来,放到配置文件中,通过"反射"找到匹配配置文件总的对 ...
- windows下的C++与cuda编译器位置
在windows下最常见的C++编译器为visual studio自带的编译器cl.exe 通常其所在目录为: C:\Program Files (x86)\Microsoft Visual Stud ...
- Hibernate的10个常见面试问题及答案
在Java J2EE方面进行面试时,常被问起的Hibernate面试问题,大多都是针对基于Web的企业级应用开发者的角色的.Hibernate框架在Java界的成功和高度的可接受性使得它成为了Java ...