单细胞分析实录(3): Cell Hashing数据拆分
在之前的文章里,我主要讲了如下两个内容:(1) 认识Cell Hashing;(2): 使用Cell Ranger得到表达矩阵。相信大家已经知道了cell hashing与普通10X转录组的差异,以及使用cellranger得到表达矩阵。
这一篇讲如何使用Seurat的HTODemux函数,CiteFuse的crossSampleDoublets函数两种方法拆分表达矩阵(混了不同来源的细胞),最后还会略微比较一下两种方法得到的结果的差异。
HTODemux
这种方法的原理我在第一篇笔记中已经讲过,感兴趣的小伙伴可以看之前的文章。主要R代码如下:
library(Seurat)
library(ggplot2)
library(tidyverse)
args <- commandArgs(TRUE)
加载R包,导入外部参数,args[1]表示样本名称,args[2]表示ensembl_ID和基因symbol对应关系的文本文件,前面得到的表达矩阵行名是ensembl_ID,为了在后续可视化的时候更省事,建议在这一步更换基因名称。
df <- read.table(paste(args[1],".mat.count.txt",sep = ""),header = T,row.names = 1) #df的行数包括基因和tag
colnames(df) <- str_replace(colnames(df),"\\.1","")
ensembl_symbol <- read.table(args[2],header = F,row.names = 1,stringsAsFactors = F)
df1 <- df[intersect(rownames(ensembl_symbol),rownames(df)),] #提取出基因表达矩阵
df2 <- df[setdiff(rownames(df),rownames(ensembl_symbol)),] #提取出tag表达矩阵
rownames(df1) <- ensembl_symbol[rownames(df1),] #更换基因表达矩阵的行名
接下来利用df2数据框的信息拆分,df2行为tag,列为cellular barcode
cellhash <- CreateSeuratObject(counts = df2,project = "cell_hashing", assay = "HTO")
cellhash <- NormalizeData(cellhash, assay = "HTO", normalization.method = "CLR")
cellhash <- HTODemux(cellhash, assay = "HTO", positive.quantile = 0.85)
最后一步就是拆分,第一篇笔记说过,positive.quantile参数表示在拟合负二项分布之后使用什么分位数来判断UMI是相对大还是相对小,默认值是0.99,实际处理时,发现这个值可能并不合理,比如最终拆分出来的有效细胞数、不同来源细胞数比例和预期差别很大,再比如从图形上看,明显不对(下文有图形说明)。
这一步之后,每一个CB都会带上一个标签,比如我的数据只有两个样本来源,标签会有这4种:Negative、tag6_tag7、tag6、tag7,前面两个表示空液滴、(跨样本的)doublet。
Idents(cellhash) <- "HTO_classification"
FeatureScatter(cellhash, feature1 = paste("hto_",rownames(cellhash)[1],sep=""),
feature2 = paste("hto_",rownames(cellhash)[2],sep = ""),slot = "counts")
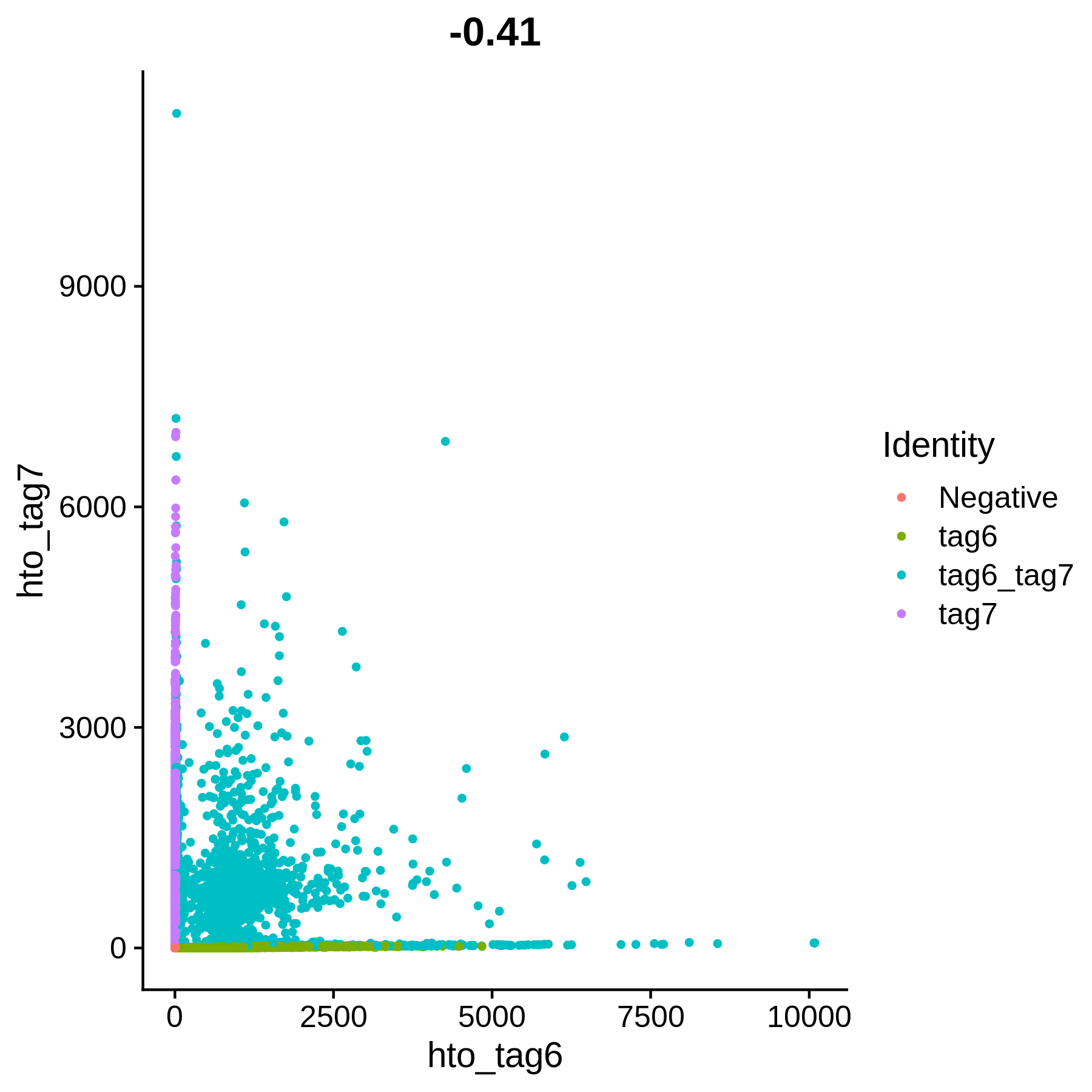
HTOHeatmap(cellhash, assay = "HTO")

上面两个图,可以用来检验拆分的质量,第一张每个点的横纵坐标表示每个CB两个tag的UMI,第二张图的每一列表示每个CB两个tag的标准化之后的表达量。
然后根据每个CB的标签提取出有效的singlet就可以了。
small_df1 <- df1[,colnames(cellhash)[cellhash$HTO_classification==rownames(cellhash)[1]]]
write.table(small_df1,paste(args[1],"_",rownames(cellhash)[1],".mat.count.txt",sep = ""),quote = F,row.names = T,col.names = T,sep="\t")
small_df2 <- df1[,colnames(cellhash)[cellhash$HTO_classification==rownames(cellhash)[2]]]
write.table(small_df2,paste(args[1],"_",rownames(cellhash)[2],".mat.count.txt",sep = ""),quote = F,row.names = T,col.names = T,sep="\t")
除了上面两种Seurat自带图形,下面两种图形也很有参考意义,代码就先不放了,如有需要可以在公众号后台小窗我。
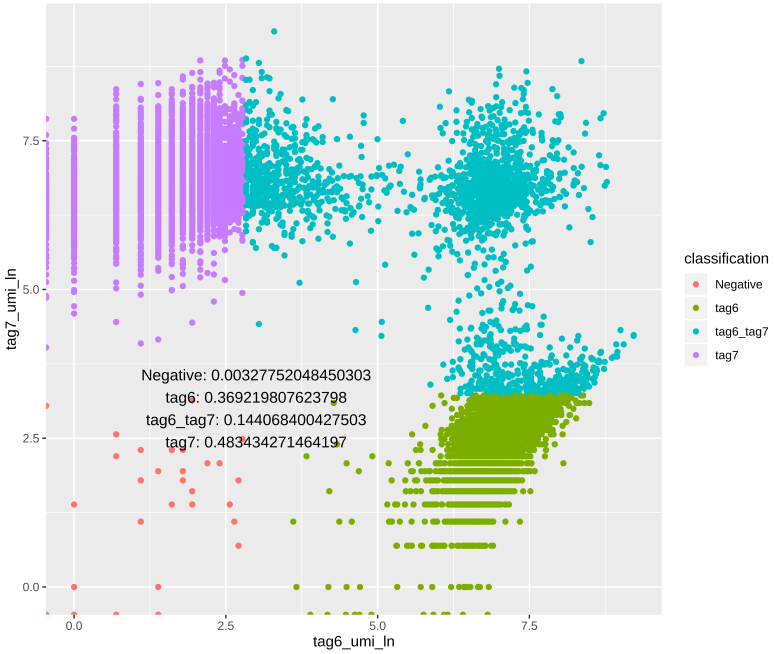

将UMI取对数之后做图,可以从另一个角度看结果,可以看到右上角被HTODemux认定为doublet的CB,像是包含了本应该是singlet的CB。我尝试过positive.quantile用默认值0.99,这种现象会更明显,所以我觉得在做这一步的时候,可以画画这个图,选择一个适中的positive.quantile值。
crossSampleDoublets
CiteFuse包在做这一步的时候,是从取对数之后的UMI矩阵开始的,分别从两个维度拟合正态分布,因此最终得到的结果在散点图上,比上一种方法更说得过去。示意图如下:

具体使用的R代码如下:
library(tidyverse)
library(ggplot2)
library(SingleCellExperiment)
library(CiteFuse)
args <- commandArgs(TRUE)
allexp <- read.table(paste(args[1],".mat.count.txt",sep = ""),header = T,row.names = 1)
colnames(allexp) <- str_replace(colnames(allexp),"\\.1","")
allexp_sce <- preprocessing(exprsMat = as.matrix(allexp)) #生成特定的对象
is.HTO <- grepl("^tag[123678]", rownames(allexp_sce)) #根据自己的tag命名修改正则表达式
allexp_sce <- splitAltExps(allexp_sce, ifelse(is.HTO, "HTO", "gene")) #给每一行加一个标签,HTO或者gene
allexp_sce=normaliseExprs(allexp_sce, altExp_name = "HTO", exprs_value = "counts",transform = c("log")) #仅针对HTO行,取对数
allexp_sce=crossSampleDoublets(allexp_sce,altExp_name = "HTO",totalExp_threshold = 10)
最后一行就是拆分关键步骤,会给每个CB一个标签,totalExp_threshold表示只会保留表达数大于10的CB。
ensembl_symbol <- read.table("/ref/10x/Ensembl_symbol_new.txt",header = F,row.names = 1,stri
ngsAsFactors = F)
df1 <- allexp[intersect(rownames(ensembl_symbol),rownames(allexp)),]
df2 <- allexp[setdiff(rownames(allexp),rownames(ensembl_symbol)),]
rownames(df1) <- ensembl_symbol[rownames(df1),]
tmp1=as.data.frame(t(df2))
tmp2=as.data.frame(allexp_sce$doubletClassify_between_label)
colnames(tmp2)="anno"
tmp2$anno=as.character(tmp2$anno)
crossSampleDoublets返回的标签不容易识别,比如1、2,还需要重新更换名称,如下
for (i in seq(1,length(rownames(tmp2)),1)) {
for (j in seq(1,length(colnames(tmp2)),1)) {
if (tmp2[i,j] == "1") {
tmp2[i,j] = colnames(tmp1)[1]
}
if (tmp2[i,j] == "2") {
tmp2[i,j] = colnames(tmp1)[2]
}
if (tmp2[i,j] == "doublet/multiplet") {
tmp2[i,j] = "doublet"
}
}
}
df_point=cbind(tmp1,tmp2)
colnames(df_point)=c("taga","tagb","anno")
这一步之后就能根据tag标签画散点图,以及提取想要的矩阵了。
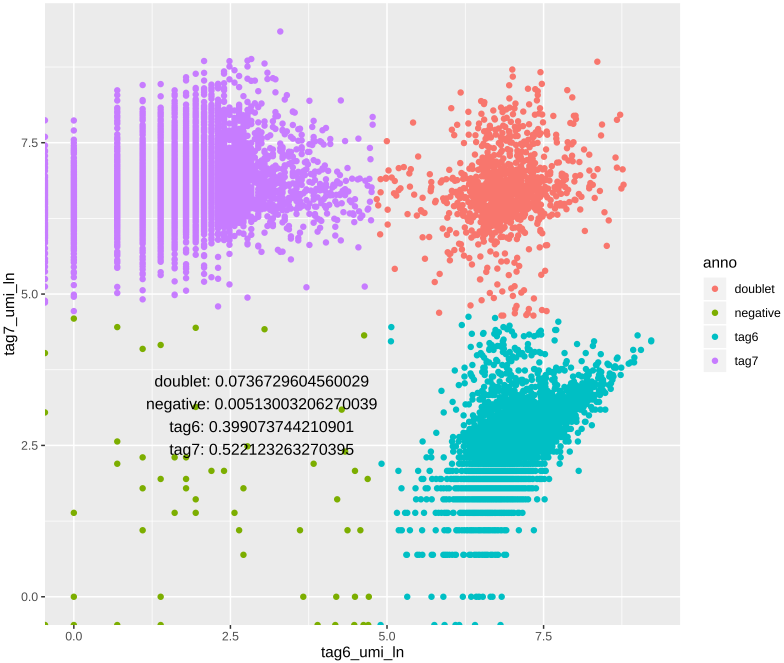
实际处理中,上面两种方法我都用了,最后选了二者交集的CB来提取矩阵(相对保险的做法)。这一步在cell hashing数据的处理中可以说是相当重要了,如果拆分质量不过关,错误地将不同来源的细胞划分到一个矩阵中,对后续分析结果影响很大。
上述代码只呈现了拆分的关键步骤,详细的画图代码没有放上来,如果需要可以在微信后台私信我。
因水平有限,有错误的地方,欢迎批评指正!

单细胞分析实录(3): Cell Hashing数据拆分的更多相关文章
- 单细胞分析实录(1): 认识Cell Hashing
这是一个新系列 差不多是一年以前,我定导后没多久,接手了读研后的第一个课题.合作方是医院,和我对接的是一名博一的医学生,最开始两边的老师很排斥常规的单细胞文章思路,即各大类细胞分群.注释.描述,所以起 ...
- 单细胞分析实录(2): 使用Cell Ranger得到表达矩阵
Cell Ranger是一个"傻瓜"软件,你只需提供原始的fastq文件,它就会返回feature-barcode表达矩阵.为啥不说是gene-cell,举个例子,cell has ...
- 【代码更新】单细胞分析实录(21): 非负矩阵分解(NMF)的R代码实现,只需两步,啥图都有
1. 起因 之前的代码(单细胞分析实录(17): 非负矩阵分解(NMF)代码演示)没有涉及到python语法,只有4个python命令行,就跟Linux下面的ls grep一样的.然鹅,有几个小伙伴不 ...
- 【代码更新】单细胞分析实录(20): 将多个样本的CNV定位到染色体臂,并画热图
之前写过三篇和CNV相关的帖子,如果你做肿瘤单细胞转录组,大概率看过: 单细胞分析实录(11): inferCNV的基本用法 单细胞分析实录(12): 如何推断肿瘤细胞 单细胞分析实录(13): in ...
- 单细胞分析实录(4): doublet检测
最近Cell Systems杂志发表了一篇针对现有几种检测单细胞测序doublet的工具的评估文章,系统比较了常见的例如Scrublet.DoubletFinder等工具在检测准确性.计算效率等方面的 ...
- 单细胞分析实录(5): Seurat标准流程
前面我们已经学习了单细胞转录组分析的:使用Cell Ranger得到表达矩阵和doublet检测,今天我们开始Seurat标准流程的学习.这一部分的内容,网上有很多帖子,基本上都是把Seurat官网P ...
- 单细胞分析实录(17): 非负矩阵分解(NMF)代码演示
本次演示使用的数据来自2017年发表于Cell的头颈鳞癌单细胞文章:Single-Cell Transcriptomic Analysis of Primary and Metastatic Tumo ...
- 单细胞分析实录(8): 展示marker基因的4种图形(一)
今天的内容讲讲单细胞文章中经常出现的展示细胞marker的图:tsne/umap图.热图.堆叠小提琴图.气泡图,每个图我都会用两种方法绘制. 使用的数据来自文献:Single-cell transcr ...
- 单细胞分析实录(18): 基于CellPhoneDB的细胞通讯分析及可视化 (上篇)
细胞通讯分析可以给我们一些细胞类群之间相互调控/交流的信息,这种细胞之间的调控主要是通过受配体结合,传递信号来实现的.不同的分化.疾病过程,可能存在特异的细胞通讯关系,因此阐明这些通讯关系至关重要. ...
随机推荐
- JZOJ 2020.10.6 【NOIP2017提高A组模拟9.7】简单无向图
简单无向图 题目 Description Input Output Sample Input 输入1: 4 2 1 1 2 输入2: 10 2 2 2 2 1 1 2 1 1 2 Sample Out ...
- day3(使用axios实现登录成功)
1.创建一个login.vue页面 1.1写页面components/Login.vue 在 src/components 下创建 Login.vue 页面 <template> &l ...
- PyQt(Python+Qt)学习随笔:Designer中PushButton按钮default、atuoDefault属性
引言 1.default.atuoDefault属性仅在父窗口为对话窗才生效,其他窗口类型设置这两个属性没有意义: 2.按钮的按压触发除了鼠标键之外,也可以使用回车键和空格键触发,这两个属性正是控制回 ...
- Monkey的使用
1.进入adb shell 环境 在Windows环境下进入DOS界面,输入adb shell 注意:adb shell服务使用的端口是5037,如果此端口被其他进程占用时,将不能正常启动adb sh ...
- Codeforces Edu Round 65 A-E
A. Telephone Number 跟之前有一道必胜策略是一样的,\(n - 10\)位之前的数存在\(8\)即可. #include <iostream> #include < ...
- Java并发编程的艺术(九)——闭锁、同步屏障和信号量
闭锁:CountDownLatch 使用场景 当前线程需要等待若干条线程执行完毕后,才能继续执行的情况. 也可以是若干个步骤执行完毕后的情况. 使用方法 初始化闭锁的时候,填入计数值,然后等待其他线程 ...
- http请求user_agent字段解析
浏览器的常见User Agent 各字段的解释 浏览器的User Agent字段令人迷惑,例如:某一版本的Chrome访问网络时,User Agent字段如下: Mozilla/5.0 (Window ...
- Python搭建调用本地dll的Windows服务(浏览器可以访问,附测试dll64位和32位文件)
一.前言说明 博客声明:此文链接地址https://www.cnblogs.com/Vrapile/p/14113683.html,请尊重原创,未经允许禁止转载!!! 1. 功能简述 (1)本文提供生 ...
- sql server的bcp指令
有时需要允许bcp指令 -- 允许配置高级选项EXEC sp_configure 'show advanced options', 1GO-- 重新配置RECONFIGUREGO-- 启用xp_cmd ...
- spring入门学习
开发步骤: 1.导入Spring开发的基本坐标 2.编写接口和实现类 3.创建Spring核心配置文件 4.在Spring核心配置文件中配置实现类 5.使用Spring的API获得Bean实例Bean ...