Bioconductor 中的 R 包安装教程
Bioconductor 是一个基于 R 语言的生物信息软件包,主要用于生物数据的注释、分析、统计、以及可视化(http://www.bioconductor.org)。
总所周知,Bioconductor 是和 R 版本绑定的,这是为了确保用户不把包安装在错误的版本上。Bioconductor 发行版每年更新两次,它在任何时候都有一个发行版本(release version),对应于 R 的发行版本。此外,Bioconductor 还有一个开发版本(development version),它对应于 R 的开发版本。
R 每年(通常是 4 月中旬)在 ‘x.y.z’ 中发布一个 ‘.y’ 版本,但 Bioconductor 每 6 个月(4 月中旬和 10 月中旬)发布一个 ‘.y’ 版本。
Bioconductor 与 R 各自对应的版本如下:(Bioconductor releases) 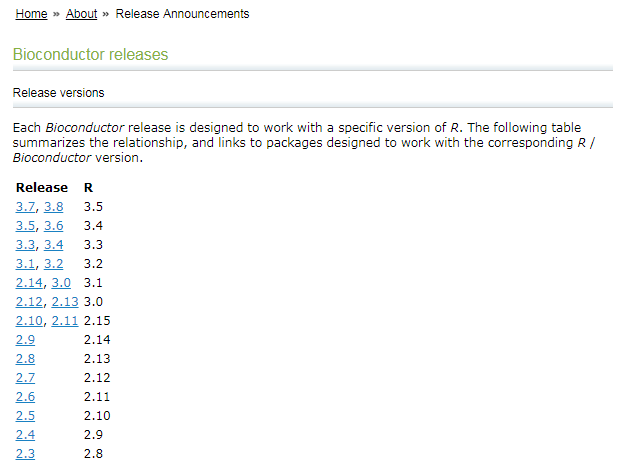
biocLite
在 R==3.5(Bioconductor-3.7 前,Bioconductor 都是通过 biocLite 安装相关的 R 包:
source("https://bioconductor.org/biocLite.R")
biocLite(pkg_name)但是,从 R-3.5(Bioconductor-3.8)起,Bioconductor 更改了 R 包的安装方式:它们通过发布在 CRAN 的 BiocManager 包来对 Bioconductor 的包进行安装和管理——通过 CRAN 安装 BiocManager,再通过这个包来安装 Bioconductor 的包。
BiocManager
安装 BiocManager 包
chooseCRANmirror()
install.packages("BiocManager")
安装 Bioconductor (or CRAN) 的 R 包
BiocManager::install(c("GenomicRanges", "Organism.dplyr"))
更新所有已经安装的 R 包到最新版本
BiocManager::install()
查看 Bioconductor 的版本
BiocManager::version()
## '3.8'
旧和意外版本的 R 包
当 Bioconductor 的包都来自同一版本时,它们的效果最佳。 使用 valid() 来查看过期(out-of-date)或意外版本(unexpected versions)的 R 包。
BiocManager::valid()
## Warning: 21 packages out-of-date; 2 packages too new
##
## * sessionInfo()
##
## R Under development (unstable) (2018-11-02 r75540)
## Platform: x86_64-pc-linux-gnu (64-bit)
## Running under: Ubuntu 18.04.1 LTS
##
## Matrix products: default
## BLAS: /usr/lib/x86_64-linux-gnu/blas/libblas.so.3.7.1
## LAPACK: /usr/lib/x86_64-linux-gnu/lapack/liblapack.so.3.7.1
##
## locale:
## [1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
## [3] LC_TIME=en_US.UTF-8 LC_COLLATE=C
## [5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
## [7] LC_PAPER=en_US.UTF-8 LC_NAME=C
## [9] LC_ADDRESS=C LC_TELEPHONE=C
## [11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
##
## attached base packages:
## [1] stats graphics grDevices utils datasets methods base
##
## other attached packages:
## [1] BiocStyle_2.11.0
##
## loaded via a namespace (and not attached):
## [1] Rcpp_1.0.0 bookdown_0.7 digest_0.6.18
## [4] rprojroot_1.3-2 backports_1.1.2 magrittr_1.5
## [7] evaluate_0.12 stringi_1.2.4 rmarkdown_1.10
## [10] tools_3.6.0 stringr_1.3.1 xfun_0.4
## [13] yaml_2.2.0 compiler_3.6.0 BiocManager_1.30.4
## [16] htmltools_0.3.6 knitr_1.20
##
## Bioconductor version '3.9'
##
## * 21 packages out-of-date
## * 2 packages too new
##
## create a valid installation with
##
## BiocManager::install(c(
## "BiocManager", "GenomicDataCommons", "GenomicRanges", "IRanges",
## "RJSONIO", "RcppArmadillo", "S4Vectors", "TCGAbiolinks", "TCGAutils",
## "TMB", "biocViews", "biomaRt", "bumphunter", "curatedMetagenomicData",
## "dimRed", "dplyr", "flowCore", "ggpubr", "ggtree", "lme4", "rcmdcheck",
## "shinyFiles", "tximportData"
## ), update = TRUE, ask = FALSE)
##
## more details: BiocManager::valid()$too_new, BiocManager::valid()$out_of_datevalid() 返回一个对象,可以查询该对象以获取有关无效包的详细信息:
> v <- valid()
Warning message:
6 packages out-of-date; 0 packages too new
> names(v)
[1] "out_of_date" "too_new"
> head(v$out_of_date, 2)
Package LibPath
bit "bit" "/home/shenweiyan/R/x86_64-pc-linux-gnu-library/3.5-Bioc-3.8"
ff "ff" "/home/shenweiyan/R/x86_64-pc-linux-gnu-library/3.5-Bioc-3.8"
Installed Built ReposVer Repository
bit "1.1-12" "3.5.0" "1.1-13" "https://cran.rstudio.com/src/contrib"
ff "2.2-13" "3.5.0" "2.2-14" "https://cran.rstudio.com/src/contrib"
>
适用的 R 包
可以使用 available() 发现适用于我们的 Bioconductor 版本的软件包; 第一个参数是可用于根据正则表达式过滤包名称,例如,可用于 Homo sapiens 的 ‘BSgenome’ 包:
avail <- BiocManager::available()
length(avail)
## [1] 16261
BiocManager::available("BSgenome.Hsapiens")
## [1] "BSgenome.Hsapiens.1000genomes.hs37d5"
## [2] "BSgenome.Hsapiens.NCBI.GRCh38"
## [3] "BSgenome.Hsapiens.UCSC.hg17"
## [4] "BSgenome.Hsapiens.UCSC.hg17.masked"
## [5] "BSgenome.Hsapiens.UCSC.hg18"
## [6] "BSgenome.Hsapiens.UCSC.hg18.masked"
## [7] "BSgenome.Hsapiens.UCSC.hg19"
## [8] "BSgenome.Hsapiens.UCSC.hg19.masked"
## [9] "BSgenome.Hsapiens.UCSC.hg38"
## [10] "BSgenome.Hsapiens.UCSC.hg38.masked"Bioconductor 中安装旧版本的 R 包
对于 R >= 3.5,Bioconductor-3.8 可以使用 BiocManager 安装相关与版本匹配的 R 包。那么使用 3.5 以下 R 版本的用户是继续使用 biocLite,还是 BiocManager,还是其他的方法安装匹配相关版本的 R 包呢?
首先,biocLite 和 BiocManager 不适用 R < 3.5。
> source("https://bioconductor.org/biocLite.R")
Bioconductor version 3.6 (BiocInstaller 1.28.0), ?biocLite for help
A new version of Bioconductor is available after installing the most recent
version of R; see http://bioconductor.org/install
> biocLite("clusterProfile")
......
Warning message:
package ‘clusterProfile’ is not available (for R version 3.4.3)
> chooseCRANmirror()
> install.packages("BiocManager")
Warning message:
package ‘BiocManager’ is not available (for R version 3.4.3)
>其次,对于 R < 3.5.0,Bioconductor 推荐使用以下命令安装相应的 R 包。
source("https://bioconductor.org/biocLite.R")
BiocInstaller::biocLite(c("GenomicFeatures", "AnnotationDbi"))Bioconductor 安装旧版本 clusterProfiler
在 Aanconda2 环境 R==3.4.3 中安装 clusterProfiler,发现 package ‘clusterProfile’ is not available (for R version 3.4.3)。
> source("https://bioconductor.org/biocLite.R")
Bioconductor version 3.6 (BiocInstaller 1.28.0), ?biocLite for help
A new version of Bioconductor is available after installing the most recent
version of R; see http://bioconductor.org/install
> biocLite("clusterProfile")
BioC_mirror: https://bioconductor.org
Using Bioconductor 3.6 (BiocInstaller 1.28.0), R 3.4.3 (2017-11-30).
Installing package(s) ‘clusterProfile’
Old packages: 'ade4', 'ape', 'backports', 'caret', ......
Update all/some/none? [a/s/n]: n
Warning message:
package ‘clusterProfile’ is not available (for R version 3.4.3)使用 BiocInstaller 安装 clusterProfiler:
> source("https://bioconductor.org/biocLite.R")
Bioconductor version 3.6 (BiocInstaller 1.28.0), ?biocLite for help
A new version of Bioconductor is available after installing the most recent
version of R; see http://bioconductor.org/install
> BiocInstaller::biocLite("clusterProfiler")
BioC_mirror: https://bioconductor.org
Using Bioconductor 3.6 (BiocInstaller 1.28.0), R 3.4.3 (2017-11-30).
Installing package(s) ‘clusterProfiler’
trying URL 'https://bioconductor.org/packages/3.6/bioc/src/contrib/clusterProfiler_3.6.0.tar.gz'
Content type 'application/x-gzip' length 4478098 bytes (4.3 MB)
==================================================
downloaded 4.3 MB
* installing *source* package ‘clusterProfiler’ ...
** R
** data
** inst
** byte-compile and prepare package for lazy loading
** help
*** installing help indices
** building package indices
** installing vignettes
** testing if installed package can be loaded
* DONE (clusterProfiler)
> packageVersion('clusterProfiler')
[1] ‘3.6.0’
本文分享自微信公众号 - 生信科技爱好者(bioitee)。
如有侵权,请联系 support@oschina.cn 删除。
本文参与“OSC源创计划”,欢迎正在阅读的你也加入,一起分享。
Bioconductor 中的 R 包安装教程的更多相关文章
- R 包安装、载入和卸载
生物上的一些包可以这样安装 source("https://bioconductor.org/biocLite.R") biocLite("affy") 一般的 ...
- Linux环境下R和R包安装及其管理
前言 R对windows使用很友好,对Linux来说充满了敌意.小数据可以在windows下交互操作,效果很好很棒.可是当我们要处理大数据,或者要在集群上搭建pipeline时,不得不面对在Linux ...
- Sublime Text 2安装汉化破解、插件包安装教程
原文地址: Sublime Text 2安装汉化破解.插件包安装教程_百度经验 http://jingyan.baidu.com/article/ff4116259b057c12e48237b8.ht ...
- windows系统mysql-5.7官方绿色版zip包安装教程
准备 下载页面:https://dev.mysql.com/downloads/mysql/ 点击 Download 按钮下载zip包到本地,解压(以我本地的解压路径是 D:\db\mysql-5.7 ...
- 如何将R包安装到自定义路径
参考 设置环境变量R_LIBS将R包安装到自定义路径 实际上是可以解决问题的, #环境变量完成以后,启动(重启)R,运行 .libPaths() 加载R包时,发现路径仍然未变成自定义的. 那么参 ...
- R包安装的正确方式
options("repos" = c(CRAN="https://mirrors.tuna.tsinghua.edu.cn/CRAN/")) if(! req ...
- Hadoop学习---Ubuntu中hadoop完全分布式安装教程
软件版本 Hadoop版本号:hadoop-2.6.0-cdh5.7.0: VMWare版本号:VMware 9或10 Linux系统:CentOS 6.4-6.5 或Ubuntu版本号:ubuntu ...
- VirtualBox扩展包安装教程|VirtualBox扩展增强包怎么安装
VirtualBox是一款功能强大的免费虚拟机软件,一般我们安装VirtualBox后要安装扩展增强包,VirtualBox扩展包包含USB2.0和USB3.0控制等支持功能,如果没有装,在使用过程中 ...
- vs中python包安装教程
vs安装python很简单,只需要在vs安装包中选择python就可以了,这里使用的python3.7: 如果有了解,都知道安装python包的指令:"pip install xxx&quo ...
- Xbox360自制系统GOD包安装教程
1.准备工作 U盘或移动硬盘一个,已下载好的GOD包,本教程用一个32G的U盘和游戏<猎天使魔女>为例. 右击U盘,属性,查看你的U盘是否为FAT32格式. 如果是FAT32格式,则可直接 ...
随机推荐
- Flink 1.0 ProgramInvocationException: Job failed ConnectException: 拒绝连接 (Connection refused)
[问题描述]:[root@hadoop1 flink-1.10.1]# bin/flink run examples/streaming/SocketWindowWordCount.jar --po ...
- Kingpin Private Browser - 隐私保护浏览器,隐身模式、广告拦截做你的私人浏览器
Kingpin Private Browser 是一个功能齐全的浏览器,隐身模式和广告拦截总是启用.它不会记住历史记录.密码或cookie.默认情况下,浏览器使用谷歌搜索(您可以在设置中将其更改为Du ...
- jason数组实现页面
<!DOCTYPE html> <html lang="en"> <head> <meta charset="UTF-8&quo ...
- ubuntu安装ch34x驱动,并安装串口调试助手
1.查看系统自带的ch34x驱动 kangxubo@kangxubo-HKNS:/lib/modules/5.19.0-38-generic/kernel/drivers/usb/serial$ ls ...
- .net core基于HttpClient实现的网络请求库
Soda.Http 基于HttpClient封装的 Http 请求库.如果有什么好想法,可以提 Issue 或者 Pr.,如果想要使用,直接在nuget搜索Soda.Http即可. Github项目地 ...
- [Java EE] java.net.SocketException: Connection reset【解决中】
1 错误描述 6:44:33.112] [DEBUG] [http-nio-9527-exec-3] [HttpClientUtil] http post url:http://bdp-gateway ...
- [Java JDK]ResultSet.next()
1 JDK [jdk1.5doc] Moves the cursor down one row from its current position. A ResultSet cursor is ini ...
- MordernC++之 auto 和 decltype
在C++11标准中,auto作为关键字被引入,可以用来自动推导变量类型,auto可以用于定义变量,函数返回值,lambda表达式等,在定义变量时可以使用auto来代替具体类型,编译器根据变量初始化表达 ...
- Docker容器网络(基本网络模型)
解析Docker的4种容器网络 默认网络模型 先介绍默认的网络模型: 安装docker后,输入ifconfig就会发现多了网卡中多了一个docker0: $ ifconfig docker0: fla ...
- [Pytorch框架] 2.1.3 神经网络包nn和优化器optm
文章目录 PyTorch 基础 : 神经网络包nn和优化器optm 定义一个网络 损失函数 优化器 PyTorch 基础 : 神经网络包nn和优化器optm torch.nn是专门为神经网络设计的模块 ...