Evaluate|GC content|Phred|BAC|heterozygous single nucleotide polymorphisms|estimate genome size|
(Evaluate):检查reads,可使用比对软件:使用SOAPaligner重新排列;采用massively parallel next-generation sequencing technology,效果很好(因为覆盖率高,精度高)
重新做有何意义:此时不需要过高的测序深度,因为用原来的read向之前assembly的基因组上比对,此时的测序深度也可以自己设定,20X以上就很好。
massively parallel next-generation sequencing technology是什么?
(GC content)检查每500bp(因为DNA片段的长度大概是500bp)滑窗的GC含量(因为CG含量应该是均一的,在这里是要检查是否均一,如果不均一可以发现错配或者该基因的结构特征),我们也发现了CG含量过高和过低值(因为只有少部分(因为它比人和狗的基因组具有相似性,但异常CG含量都少)片段有这种异常),熊猫与人和狗记忆中的差异性在于:熊猫assembly中缺失了一部分CG含量高的基因。所以可以知道,本次assembly并不被GC含量异常所严重影响。
500-bp non-overlapping sliding windows:用来分区域检查组装成果,这里500bp为一个单位,检查该单位中的碱基含量等,碱基分布是否均匀(一般正常是GC含量占40%,因为碱基互补配对原则),基因组中存在多少比例的高GC区域等。据此,可在一定程度上推测物种基因组结构特征,组装中是否存在明显的错配,或判断测序数据中是否存在其他物种污染等
GC biased non-random sampling:因为GC的氢键有三条,所以该样本在打碎时,GC氢键不易打断,所以这是可选择的采样。
sufficient for de novo assembly:虽然存在错误,但是可以容忍
(compare with GeneBank):除Y染色体性别决定区的基因外,其余26个基因的比对成功率很高。特别是RPS15(核糖体)基因,这说明assembly的coverage and completeness很好,(全面性补充:因为核糖体基因自身性质,所以存在多拷贝和拥有重复序列的片段)
SRY sex:sex-determining region of Y-chromosome,Y染色体性别决定区
为什么核糖体存在多拷贝和拥有重复序列的片段?因为核糖体要快速组装,所以多拷贝和重复序列是最快的。
(assess the large-scale and local assembly accuracy of the scaffolds):针对scaffold,组装人工细菌染色体,利用人工细菌染色体拷贝了一个scaffold( large-scale),查看单碱基错配和插入缺失情况(local assembly accuracy),这些情况是由于未知的SNP情况(因为在BAC上的depth高(可靠)并且差异处phred高(Phred)的因为这是排除了已注释的SNP和嵌合体的条件下做的,所以只能是未知的SNP。
Phred计算许多与波峰大小和分辨率相关的参数,根据这些参数,从一个巨大的查询表中找出碱基质量得分,这里质量得分高,则证明可靠性。
杂合SNP:heterozygous single nucleotide polymorphisms:单碱基突变(SNP),发生在成对两条染色体上,所以是杂合现象,即染色体1和染色体2是成对染色体,染色体1 上某处碱基是A与它互补的链上碱基是T,但是染色体2上相对应的染色体相应处本来应该是A,但是由于SNP所以变成了G,则与之对应的链上碱基为C,则该处序列,四种碱基都存在。
嵌合体:杂合体是嵌合程度最大的嵌合体,就是每个亲代动物的遗传基因各占一半,不是杂合动物,则基因占比不定。
(BACs) independently using Sanger sequencing technology:是因为比较准,用sanger测长序列比较准,但是贵。
(genome coverage of the assembled contigs and scaffolds)使用17base寡核苷酸,依据其出现频率看深度,得到深度后,综合(sequencing depth+size ratio of syntenic blocks+C-values)因为存在的序列错误(repeat)所以我们应该得到比现有大小更小的基因组大小
sequencing depth:
Reference:http://blog.sciencenet.cn/blog-3406804-1162384.html
“如下所示,使用某物种的二代测序数据计算k-mer(选取k-mer长度17),最后可得到一个k-mer频数分布表(下图左图),第一列为k-mer深度,即各k-mer的出现频数;第二列为出现该频数的k-mer片段总数。下图右图为k-mer频数分布图,使用左图的统计表数据所绘制,图中横坐标为各k-mer的出现频数(Frequency),纵坐标为出现该频数的k-mer片段总数(Number)。


可以发现原始图中,最左侧(Frequency = 1、2等起始位置处)出现了很高的值,表明测序结果中存在大量的k-mer仅出现了1-2次,这个在k-mer频数统计表中也可轻易发现。这是因为在实际的二代测序数据中,由于测序错误(如Illumina测序平台的平均错误率约1%)的存在会引入许多带有错误碱基的reads,将这些reads打断成长度K的k-mer后,会产生许多错误的k-mer。由于测序错误带来的碱基类型是随机的,因此可知这些错误k-mer的出现频数很低,但总数目却非常的多。因此在上图中,低频数的k-mer数目占很大的比例,即在Frequency = 1、2等起始位置处出现很高的k-mer数目,使得图中曲线峰值很难分辨;为了增强曲线的可读性,可选择在作图时屏蔽掉曲线最左侧区域。当然也不排除一些真实的核酸序列,由于其碱基组成具有特异性且其只被测序测到了一次,将该序列截断为一定长度的k-mer之后这些k-mer只出现了唯一一次。但是相较于测序错误所产生的k-mer数量,后面这种情况所产生的k-mer数量基本上可忽略了,除非在很低深度的测序模式下。
此外,我们也可轻易看到,出现次数为几百上千次的k-mer数量其实很少。尽管在统计时不可丢弃这些出现频数很高但总体数量很少k-mer,但只是作图展示k-mer频数分布的话,是无需展示这些高频数深度的k-mer的,以便增强曲线的可读性(一些k-mer分析软件会统计至很高的k-mer频数深度,如10000,事实上在绘制k-mer曲线图时用不到这么多,视情况加以取舍)。
通常情况下,会考虑将低频数和高频数的数据屏蔽掉,屏蔽频数区间根据实际情况而定。屏蔽Frequency = 1、2等起始位置处以及Frequency > 500或1000等高频深度的数据后,峰值即可呈现出,结果示例如下图所示(使用数据同上,只展示5 ≤ Frequency ≤ 500的区域)。此时,在不考虑测序错误率、基因组的杂合度和重复度的情况下,逐碱基取k-mer,则k-mer曲线在理想状态下服从泊松分布。
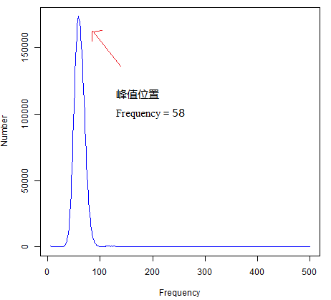
上述我们获得了k-mer频数统计结果,接下来可以根据这个统计结果初步估算测序物种基因组特征。其中,k-mer分析估算基因组大小的原理如下。
从reads中逐碱基取出的所有k-mer能够遍历整个基因组。根据Lander waterman算法,基因组大小(G)满足如下公式:
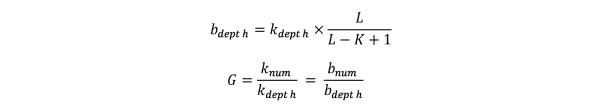
其中,L为reads平均长度,K为k-mer长度;knum为所有的k-mer总个数,kdepth为k-mer频数的期望深度(即k-mer曲线中主峰对应的横坐标位置);bnum为测序reads覆盖碱基的总个数,bdepth为覆盖碱基的期望深度。”
在这里,我们即可根据测序数据中的k-mer频数分布统计结果,大致估算出物种基因组大小了。
修正:覆盖度&深度:覆盖深度常常决定了特定碱基位置的变异发现是否具有某种水平的可信度
size ratio of syntenic blocks:共线区域尺寸比在这里对于估算基因组大小有何作用?
k-mer:有这么个reads(当然实际比这个长):AACTGACTGA.如果k-mer的k为3的话,我们可以将其切割为AAC ACT CTG TGA GAC ACT CTG TGA
(refine our estimate of the panda genome size)狗和熊猫的保守区98%,且熊与熊猫的染色体组型相似(但熊基因组大小不可知,狗基因组大小可知同时狗和熊C-value可知,比较狗和熊C-value,可知道熊基因组大小小于狗。),推断其基因组大小相似,所以我们确认基因组大小是2.4Gb。因此,使用该基因组大小得到contig和scaffold 的coverage高。
C-value:(图像中单倍体基因总含量)值,与基因组总含量有成正比吗?C值矛盾
Evaluate|GC content|Phred|BAC|heterozygous single nucleotide polymorphisms|estimate genome size|的更多相关文章
- SNP (Single Nucleotide Polymorphism), SNV ( single nucleotide variants ) , Indel (insertion-deletion) 的区别
SNP (Single Nucleotide Polymorphism):强调在一个群体中具有一定频率的变异,一般为二态性.比如G→C SNV ( single nucleotide variants ...
- 05 Computing GC Content
Problem The GC-content of a DNA string is given by the percentage of symbols in the string that are ...
- 单核苷酸多态性SNP(single nucleotide polymorphism)
定义 主要指基因组水平上由单个核苷酸的变异所引起的 DNA 序列多态性. 在基因组水平上由单个核苷酸的变异所引起的DNA序列多态性.即:在不同个体的同一条染色体或同一位点的核苷酸序列中,绝大多数核苷酸 ...
- 什么是侧翼区(flanking region)和侧翼区单核苷酸多态性(Flanking SNPs)
侧翼区(flanking region) 根据维基定义:The 5' flanking region is a region of DNA that is adjacent to the 5' end ...
- SNPs & MAF
SNPs,全称是single nucleotide polymorphisms,SNPs等位基因频率的容易估计.采用混和样本估算等位基因的频率是种高效快速的策略.该策略的原理是:首先选择参考样本制作标 ...
- SNP(单核苷酸多态性)准确性的验证,你造吗?
SNP(单核苷酸多态性)准确性的验证,你造吗? [2016-12-12] SNP(全称Single Nucleotide Polymorphisms)即单核苷酸多态性,主要是指在基因组水平 ...
- ADNI数据和样例
ADNI临床数据集: 由各个学科的临床信息组成,包括招募.人口统计特征.体格检查和认知评估数据 所收集的临床数据: 基因数据: ILLUMINA SNP基因分型检测 ADNI的一个关键目标就是为研究人 ...
- ADNI数据集相关概念整理
数据类型 临床 遗传 MRI图像 PET图像 生物样本 临床 ADNI临床数据集包括关于每个受试者的临床信息,包括招募,人口统计学,身体检查和认知评估数据.可以将整套临床数据作为逗号分隔值(CSV)文 ...
- DNA拷贝数变异CNV检测——基础概念篇
DNA拷贝数变异CNV检测——基础概念篇 一.CNV 简介 拷贝数异常(copy number variations, CNVs)是属于基因组结构变异(structural variation), ...
随机推荐
- STL——stack
首先,堆栈是一个线性表,插入和删除只在表的一端进行.这一端称为栈顶(Stack Top),另一端则为栈底(Stack Bottom).堆栈的元素插入称为入栈,元素的删除称为出栈.由于元素的入栈和出栈总 ...
- Exists 方法
public void ExistsMethodDemo() { string userId = "123"; string userName = "admin" ...
- 51nod 1449 砝码称重【天平/进制】
题意: 给你w,n,问你在w^0,w^1,w^2...各种一个,问你能不能用这些砝码和重量为m的东西放在天平上使得天平平衡: 思路: 这个很容易联想到进制: 如果把m放在是一边的话,其实对于砝码就是纯 ...
- PTA 4-4 先序输出叶结点 【基础题】
//二叉树的叶结点:度为0的结点. void PreorderPrintLeaves( BinTree BT ) { if(BT==NULL) //如果传下来根节点就是空,直接返回: return; ...
- 2013 Noip提高组 Day2
3288积木大赛 正文 题目描述 春春幼儿园举办了一年一度的“积木大赛”.今年比赛的内容是搭建一座宽度为n的大厦,大厦可以看成由n块宽度为1的积木组成,第i块积木的最终高度需要是hi. 在搭建开始之前 ...
- P3270 [JLOI2016]成绩比较(拉格朗日插值)
传送门 挺神仙的啊-- 设\(f[i][j]\)为考虑前\(i\)门课程,有\(j\)个人被\(B\)爷碾压的方案数,那么转移为\[f[i][j]=\sum_{k=j}^{n-1}f[i-1][k]\ ...
- 洛谷 P1712 [NOI2016]区间(线段树)
传送门 考虑将所有的区间按长度排序 考虑怎么判断点被多少区间覆盖,这个可以离散化之后用一棵权值线段树来搞 然后维护两个指针$l,r$,当被覆盖次数最多的点的覆盖次数小于$m$时不断右移$r$,在覆盖次 ...
- perl 安装LOG4perl 模块
环境信息 ubuntu 12.04 64位 桌面版 Log-Log4perl 的介绍网址:http://search.cpan.org/~mschilli/Log-Log4perl-1.49/lib/ ...
- 要单独拿出来讲的a标签
a标签的属性 href属性赐予a标签力量:href属性指定要通过a标签借助浏览器请求的资源,可以是图片.视屏.网站.音频等.不加herf属性的a标签就是一个没有任何特殊样式和功能的文本容器. targ ...
- get 和 post 请求的区别(转)
转自 http://www.cnblogs.com/hyddd/archive/2009/03/31/1426026.html http://www.nowamagic.net/librarys/ve ...